


Consejo Genético
Índice de navegación
Cáncer de Mama y Ovario Hereditario
1.- Introducción:
Aproximadamente un 7% de los cánceres de mama y un 11-15% de los cánceres de ovario se consideran hereditarios, debido a una mutación genética heredada de uno de los padres.
BRCA1 y BRCA2 son los genes que se asocian con una mayor proporción de casos al cáncer de mama y ovario hereditario, en concreto el cáncer de ovario seroso de alto grado. Se han descrito otros genes de alta penetrancia asociados al cáncer de mama: TP53 (síndrome de Li-Fraumeni), PTEN (síndrome de Cowden), STK11 (síndrome de Peutz-Jeghers) o PALB2; otros genes de moderada penetrancia como ATM, CHEK2 y NBN. Y otros genes asociados al cáncer de ovario: genes MMR, RAD51C, RAD51D y BRIP1.
2.- Genes de alta penetrancia BRCA1/BRCA2 y PALB2:
El gen BRCA1 está localizado en el brazo largo del cromosoma 17 (17q21) y el gen BRCA2 en el cromosoma 13 (13q12). Las proteínas BRCA1 y BRCA2 actúan en las vías de reparación del ADN y su inactivación mediante mutación provoca indirectamente la aparición del tumor por acumulación de mutaciones en otros genes reguladores directos del ciclo celular.
El gen PALB2 (partner and localizer of BRCA2) es un gen relacionado con la anemia de Fanconi. Codifica una proteína que posee un papel esencial en el mantenimiento del genoma, reparando la rotura de la doble cadena de ADN. La función de la proteína PALB2 se desarrolla conjuntamente con BRCA1 y BRCA2 en la misma vía de reparación del ADN.
3. Genes de moderada penetrancia que aumentan el riesgo de cáncer de mama
Los siguientes genes de moderada penetrancia aumentan el riesgo de cáncer de mama en menor medida.
3.1. Gen ATM
ATM o gen ataxia telangiectasia mutado, es un gen localizado en el brazo largo del cromosoma 11, codifica la proteína ATM serina/treonina quinasa que está implicada en la regulación de los procesos de control de la división celular y en la reparación de daños sufridos por la molécula de ADN.
El gen ATM puede aumentar el riesgo de cáncer de mama. En un meta-análisis se estimó un riesgo relativo de 2.8 (90% IC, 2.2-3.7, p>0.001). Otros análisis muestran un 1% de mutaciones en mujeres con cáncer de mama. Y un estudio holandés de mujeres con cáncer de mama a edad temprana detectó un 8.5% de mutaciones del gen ATM.
3.2 Gen CHEK2
El gen CHEK2 (cell cycle checkpoint kinase 2) es otro de los genes identificados que aumenta el riesgo de cáncer de mama. Regula los puntos de control del ciclo celular y la apoptosis en respuesta al daño en el ADN, particularmente roturas del ADN de doble cadena.
En un estudio que no detectó mutaciones en BRCA1/2 en mujeres con una fuerte historia familiar de cáncer de mama/ovario se detectó en el 5% de estas. El riesgo acumulado de cáncer de mama en mujeres con mutación de CHEK2 e historia familiar de cáncer de mama se ha estimado entre el 28 y el 37%, siendo mayor en mujeres con antecedentes de cáncer de mama familiar. El riesgo estimado es de 3.0 (90% IC, 2.6-3.5).
Se ha estudiado la asociación de la variante truncada 1100delC con el riesgo de cáncer de mama en mujeres no seleccionadas por su historia familiar, detectando un riesgo relativo de 2.34 en el estudio del consorcio europeo y australiano. Sin embargo, esta mutación no se ha comprobado que esté presente en la población española de cáncer de mama familiar.
3.3. Gen NBN
El gen NBN es responsable de sintetizar la proteína nibrina. La nibrina es una proteína asociada con la reparación de roturas de doble hélice del ADN que ocasionarían serios daños al genoma.
Las mujeres con mutaciones heterocigotas en este gen tienen un aumento del riesgo de cáncer de mama con un RR 3.1. Un meta-análisis incluyendo 7 estudios mostró un aumento significativo con la variante 657del5 (RR 2.42).
4. Genes de alta y moderada penetrancia que aumentan el riesgo de cáncer de ovario
Hay otros genes de la familia de la proteína RAD51, involucrados en la recombinación homóloga y reparación del ADN como RAD51C y RAD51D, o el gen BRIP1, que aumentan el riesgo de CO, pero no hay evidencia clara sobre el aumento del riesgo de CM.
En la población general, el riesgo de CO, trompa de Falopio o primario peritoneal es del 1,5%. Las mutaciones en los genes BRCA1/2, RAD51C, RAD51D y BRIP1 incrementan más de 5 veces el riesgo de desarrollar CO en mujeres portadoras de alteraciones en estos genes en comparación con la población general.
4.1. RAD51C y RAD51D
RAD51 es una proteína de 339 aminoácidos que desempeña un papel importante en la recombinación homóloga del ADN. La familia de genes RAD51, que incluye los genes RAD51C y RAD51D, está involucrada en la reparación del ADN mediante la interacción con los genes BRCA1 and BRCA2. En diferentes estudios, se ha concluido que RAD51C es un gen de predisposición al cáncer de ovario, y sus mutaciones se pueden encontrar en el 1% de pacientes afectas de CO. En mujeres con mutaciones en RAD51C el riesgo de CO es del 11% a los 80 años. En mujeres portadoras de mutación en el gen RAD51D el riesgo de CO a los 80 años es de un 13%.
4.2. BRIP1
BRIP1 es una proteína del grupo de anemia de Fanconi cuya función es la reparación de ADN de doble cadena. Esta proteína también parece ser importante en el CO, donde parece actuar como supresor de tumores. Las mutaciones en BRIP1 se asocian con un riesgo de hasta el 5.8% a los 80 años de CO.
5. Criterios para remitir a la unidad de consejo genético en cáncer:
La complejidad y extrema laboriosidad del estudio de ambos genes y la escasa prevalencia de mutaciones en la población hacen inviables los análisis poblacionales.
Los criterios clínicos para indicar un estudio de los genes relacionados con el cáncer de mama/ovario hereditario están basados en la historia personal y familiar (se pueden consultar en “SEOM clinical guidelines in hereditary breast and ovarian cancer (2019)”. PMID:31889241. DOI:10.1007/s12094-019-02262-0)
6. Riesgo de cáncer en mutaciones BRCA1/BRCA2:
6.1. Riesgo de cáncer de mama y ovario en portadores de mutaciones BRCA1/BRCA2
La penetrancia, definida como la probabilidad de que un portador de una mutación en BRCA1/2 desarrolle un cáncer a lo largo de su vida, suele expresarse como el riesgo acumulado de cáncer a los 70 años. La penetrancia deriva del grado de agregación familiar de cáncer.
En un meta-análisis realizado sobre la penetrancia de BRCA1 y BRCA2, a partir de los principales estudios publicados, se recoge que el riesgo acumulado de cáncer de mama a los 70 años es del 57% (95% IC, 47 a 66%) para portadores de mutación en BRCA1 y del 49% (95% IC, 40 a 57%) en BRCA2; en cuanto al cáncer de ovario seroso de alto grado se estima un riesgo acumulado a los 70 años del 40% (95% IC, 35 a 46%) para portadores de mutación en BRCA1 y del 18% (95% IC, 13 a 23%) en BRCA2.
En un estudio publicado en 2017 con una cohorte prospectiva de 6036 mujeres portadoras de mutación en el gen BRCA1 y 3820 en el gen BRCA2, el riesgo en portadoras de mutación BRCA1 de CM fue del 78% (95% IC 65% a 79%) y de CO del 44% (95% IC 36% a 53%), mientras que en portadoras de mutación en BRCA2 el riesgo de CM fue del 69% (IC 95% 61 a 77%) y de CO del 17% (95% IC 11 a 25%). La incidencia aumenta desde los 30 a 40 años en las portadoras de mutación en BRCA1 y de los 40 a 50 años para BRCA2, y se mantiene elevado a lo largo de toda la vida. El carcinoma ductal in situ de mama (CDIS) también debe considerarse como uno de los tumores que se asocian a mutaciones en estos genes. Aparece a una edad más temprana que en la población general, unos 12 años antes.
Respecto al riesgo de CM en los hombres es mayor en los portadores de mutación en BRCA1/2 respecto a la población general. La probabilidad de desarrollar CM en portadores de mutación en BRCA2 es del 8%, y del 1% en los portadores de mutación en BRCA1. En la población general el riesgo de CM es del 0.1%.
6.2. Riesgo de otros tumores en portadores de mutación BRCA1/2
Los portadores de mutaciones en BRCA1 presentan además de un riesgo elevado de CM y de CO riesgo de otros tumores. En el estudio del Breast Cancer Linkage Consortium (2002), los portadores de mutación BRCA1 presentaban un aumento del riesgo de cáncer de páncreas (RR=2,26), carcinoma de endometrio (RR=2,65), cáncer de cérvix (RR=3,72), y de cáncer de próstata en menores de 65 años (RR=1,82) (el riesgo de cáncer de próstata en mayores de 65 años no estaba aumentado).
Respecto a los portadores de mutaciones en BRCA2 también se ha documentado un aumento del riesgo de otros tipos de tumores, como cáncer de próstata. En uno de los estudios con más mujeres realizado del Breast Cancer Linkage Consortium (3728 individuos con mutación, 681 de ellos con cáncer de mama/ovario, entre 173 familias con cáncer de mama/ovario y mutación en BRCA2), estudiaron la frecuencia de otros cánceres en los familiares de 1er grado, encontrando un aumento del riesgo estadísticamente significativo en el cáncer de próstata (RR=4,65), cáncer de páncreas (RR=3,51), cáncer de vesícula biliar y conductos biliares (RR=4,97), estómago (RR=2,59), y melanoma maligno (RR=2,58); el riesgo de cáncer de próstata en hombres menores de 65 años es de 7,3. Por último, mutaciones en este gen aumentan la predisposición al cáncer de mama en varones. Es frecuente que las familias de alto riesgo con casos de cáncer de mama en varones posean mutaciones en BRCA2.
Con los datos de estudios actuales podemos resumir el riesgo de tumores asociados a BRCA1/2:
Riesgo de tumores ginecológicos:
- Cáncer en las trompas de Falopio. Un 50% de los tumores serosos en portadoras de mutaciones BRCA1/2 tienen un origen distal a las trompas de Falopio. El riesgo en la población general es del 0.2%. En un estudio retrospectivo de 108 mujeres con carcinoma en las trompas, un 33% eran portadoras de mutación BRCA1/2. Además, en mujeres portadoras es más frecuente antes de los 60 años vs no portadoras (40.3 vs 17.4%).
- Tumor primario peritoneal (es un tumor maligno poco frecuente de la cavidad peritoneal de origen extra-ovárico, con clínica e histología similar a los estadios avanzados del carcinoma seroso de alto grado de ovario). La probabilidad de desarrollo en portadoras de mutación es del 1.3%.
- Tumores de endometrio. No parece que esté aumentado en mujeres portadoras de mutación en los genes BRCA1/2. El aumento del riesgo parece estar relacionado con el tratamiento con tamoxifeno.
Cáncer de páncreas
Como se ha comentado previamente el riesgo de cáncer de páncreas es mayor en portadores de mutaciones BRCA, entre el 1 y 4.9% para BRCA1 y BRCA2 respectivamente. La mediana de edad en portadores de mutación en BRCA2 es de 67 años en los hombres y de 59 años en las mujeres.
Cáncer de próstata
En hombres portadores de mutación en BRCA2, el riesgo de cáncer de próstata es entre 5 y 9 veces el de la población general a los 65 años, lo que significa un riesgo del 33%. También se ha descrito que aparece de forma más precoz y con una mayor agresividad.
Riesgo de otros tumores
Cáncer de colon. Los datos sobre el aumento del riesgo de cáncer de colon en estos pacientes no son consistentes. Algunos estudios describen el aumento del riesgo en BRCA1, pero otros estudios no confirman esta asociación.
Melanoma. Se ha descrito asociación entre mutaciones en BRCA2 y riesgo de melanoma, pero este riesgo no está bien caracterizado. No se ha visto una clara asociación a mutaciones en BRCA1.
Tumores de estómago y vías biliares. Parece que hay aumento del riesgo de estos tumores, pero no ha sido cuantificado.
7. Riesgo de cáncer en mutaciones PALB
Las mutaciones en este gen aumentan el riesgo de cáncer de mama. Un meta-análisis de 3 estudios estimó un riesgo relativo (RR) de 5.3 (90% IC, 3.0-9.4). El riesgo de cáncer de mama asociado a mutaciones en PALB2 aumenta con la edad, con un riesgo del 13-21% a los 50 años y del 44-63% a los 80 años de edad. Este riesgo aumenta con el número de familiares afectos, de modo que el riesgo de cáncer de mama a los 70 años si no hay familiares con cáncer de mama es del 33% comparado con el 58% en las que tienen al menos 2 familiares con cáncer de mama.
El riesgo de cáncer de ovario a lo largo de la vida en portadoras de mutación en PALB2 es del 3-5% a lo largo de la vida. Las medidas de seguimiento se deberán adoptar según la historia familiar.
8. Medidas de reducción de riesgo tras la detección de mutación en BRCA
Aunque no existe un nivel de evidencia alto, se recomienda a las mujeres con alto riesgo de cáncer de mama reducir la ingesta calórica total, evitar la obesidad, realizar ejercicio físico con regularidad y moderar el consumo de alcohol.
8.1.- Seguimiento:
8.1.1.- Cribado del cáncer de mama:
La vigilancia del cáncer de mama incluye la autoexploración mamaria mensual indicada en mujeres mayores de 18 años, la exploración mamaria clínica cada 6 meses a partir de los 25 años, la mamografía anual con o sin ecografía mamaria en mujeres mayores de 30 años y hasta los 75 años y la resonancia magnética mamaria anual en mujeres mayores de 25 años y hasta los 70 años.
8.1.2.- Cribado de cáncer de ovario:
En mujeres portadoras de mutación BRCA se recomienda realizar una exploración ginecológica con ecografía transvaginal y la determinación sérica del marcador tumoral CA 125, cada 6-12 meses desde los 30 años.
8.1.3.- Seguimiento en varones portadores de la mutación BRCA:
En varones portadores de la mutación BRCA el riesgo de desarrollar carcinoma de mama es menor. Por ello, tan sólo se recomienda la autoexploración mamaria, y en caso de detectarse alguna anomalía se realizará una mamografía con o sin ecografía mamaria.
Se recomienda realizar el cribado de carcinoma de próstata mediante tacto rectal y la determinación sérica del marcador tumoral PSA a partir de los 40 años.
8.1.4.- Cribado de otros tumores:
En mujeres y hombres portadores de mutación en BRCA1 o BRCA2 se indicará el cribado de otros tumores dependiendo de la historia familiar.
8.2.- Quimioprevención:
8.2.1.- Quimioprevención del cáncer de mama:
En el momento actual no disponemos de evidencia científica para recomendar un tratamiento para reducir el riesgo de cáncer de mama en mujeres portadoras de mutación en BRCA sin enfermedad.
8.2.2.- Quimioprevención del carcinoma de ovario:
Los anticonceptivos orales han demostrado disminuir en un 50% el riesgo de desarrollar cáncer de ovario en mujeres portadoras de la mutación BRCA. Aunque, potencialmente podrían causar un ligero incremento del riesgo de cáncer de mama en portadoras de la mutación BRCA1.
8.3.- Cirugía reductora de riesgo en portadoras de mutaciones BRCA1/2:
Constituye la estrategia más efectiva de que disponemos en la actualidad para disminuir el riesgo de cáncer de mama y de ovario, logrando una reducción de riesgo superior al 90% en las mujeres portadoras de mutaciones de los genes BRCA.
8.3.1.- Cirugía reductora de riesgo de cáncer de mama:
La mastectomía bilateral reduce el riesgo de desarrollar carcinoma de mama en un 90% en mujeres portadoras de mutación en BRCA.
La mastectomía subcutánea realiza una exéresis del tejido glandular, y a diferencia de la mastectomía simple preserva la totalidad de la piel de la mama, incluidos la areola y el pezón. La mastectomía subcutánea deja como mínimo un 5% de parénquima mamario, fundamentalmente en la zona retroareolar y en la prolongación axilar, tejido que es teóricamente susceptible de cancerización.
No existe unanimidad en el modelo quirúrgico a seguir para la prevención de mujeres sanas con alto riesgo de padecer cáncer de mama. Generalmente se realiza una mastectomía bilateral profiláctica con reconstrucción inmediata.
8.3.2- Cirugía reductora de riesgo de cáncer de ovario:
La salpingo-ooforectomía bilateral en mujeres con mutación en BRCA1 entre los 35-40 años y en mujeres con mutación en BRCA2 entre los 40-45 años, que hayan cubierto sus deseos reproductivos, es la alternativa más eficaz, con una reducción del 80% del riesgo de cáncer de ovario.
Sin embargo, queda un 5% de riesgo de carcinoma peritoneal primario.
Tras la ooforectomía deben controlarse los efectos de la menopausia precoz en su repercusión sobre la osteoporosis y el perfil lipídico.
9. Medidas de reducción de riesgo tras la detección de mutación en PALB2
En las pacientes con mutaciones germinales del gen PALB2, se recomienda seguimiento mediante mamografía anual desde los 30 años, ya que a esta edad aumenta el riesgo y considerar RNM mamaria anual.
No hay datos sobre el beneficio de reducir el riesgo mediante mastectomía bilateral, por lo que se debe tener en cuenta en función de los antecedentes familiares.
El riesgo de cáncer de ovario no está claro por lo que no hay suficiente evidencia para recomendar cirugía preventiva.
PALB2 se asocia con la anemia de Fanconi heredada de forma autosómica recesiva por lo que hay que discutir las diferentes opciones reproductivas.
10. Medidas de seguimiento en mujeres con mutaciones en genes de moderada penetrancia que aumentan el riesgo de cáncer de mama.
10.1. Mutaciones en el gen ATM
Se recomienda seguimiento mediante mamografía anual desde los 40 años y considerar RNM mamaria según la historia familiar de cáncer de mama.
No hay datos sobre el beneficio de reducir el riesgo mediante mastectomía bilateral, por lo que se debe tener en cuenta con los antecedentes familiares.
Recordar la asociación a la ataxia-telengiectasia de forma recesiva por lo que hay que discutir las diferentes opciones reproductivas.
10.2. Mutaciones en el gen CHEK2
Se recomienda seguimiento mediante mamografía anual desde los 40 años y considerar RNM mamaria anual según la historia familiar.
No hay datos sobre el beneficio de reducir el riesgo mediante mastectomía bilateral, por lo que se debe tener en cuenta con los antecedentes familiares.
10.3. Mutaciones en el gen NBN
Se recomienda seguimiento mediante mamografía anual desde los 40 años, y considerar RNM mamaria anual.
No hay datos sobre el beneficio de reducir el riesgo mediante mastectomía bilateral, por lo que se debe tener en cuenta con los antecedentes familiares.
11. Medidas de seguimiento y cirugías reductoras de riesgo en mujeres con otras mutaciones que aumentan el riesgo de cáncer de ovario.
11.1. Mutaciones en el gen RAD51C y RAD51D
No existen guías de manejo de riesgo de CO en portadoras de RAD51C ni de RAD51D por ello es muy importante adaptarlas a la historia personal y familiar de CO. Debemos tener en cuenta que el diagnóstico de CO en portadoras de mutación en RAD51C generalmente es en mujeres postmenopáusicas, por lo que se puede considerar realizar la Salpingo-Ooforectomia Bilateral Profiláctica (SOBP) una vez la mujer haya alcanzado la menopausia natural. En el caso del gen RAD51D, la opción de SOBP puede considerarse a partir de los 45-50 años.
11.2. Mutaciones en el gen BRIP1
Es importante reseñar que la edad media al diagnóstico de CO en mujeres portadoras de una mutación en BRIP1 es de 63.8 años y un 93% de los casos se diagnostican a edades mayores de los 50 años. Actualmente, solo se recomiendan controles ginecológicos regulares y SOBP a partir de los 50-55 años.
12. Alternativas en mujeres de alto riesgo de carcinoma de mama hereditario tras un resultado genético no informativo
Se aconseja valorar el riesgo según un modelo probabilístico (ej. CanRisk: (https://canrisk.org). Según las guías de la American Cancer Society (ACS) se debe ofrecer vigilancia con RNM mamaria a aquellas mujeres que presenten un riesgo de cáncer de mama mayor del 20-25%, calculado según los modelos de predicción. Se recomienda mamografía y RNM anual iniciando unos 5 años antes de la edad más precoz del cáncer de mama diagnosticado en la familia.
El seguimiento ginecológico específico no es necesario en familiares sin mutación BRCA y sin antecedentes familiares de carcinoma de ovario.
No se recomienda la cirugía reductora de riesgo de carcinoma de mama.
Tabla 1: Criterios clínico·patológicos de alto riesgo de síndrome de cáncer de mama y ovario hereditario
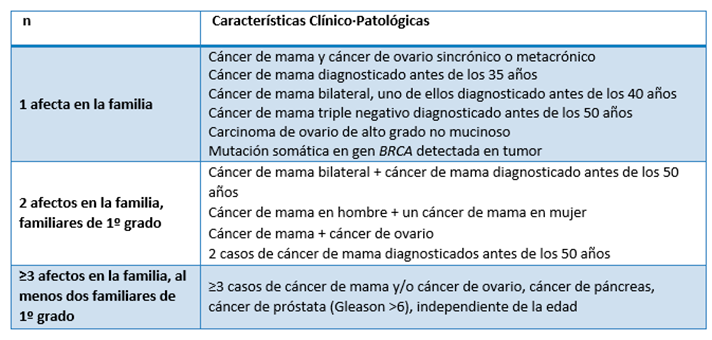
Familiares de primer grado son: madre, hermanas, hijas. No considerar a los hombres al contabilizar grado de parentesco

