


Consejo Genético
Índice de navegación
AUTOR: Dr. Rafael Morales Chamorro
Revisado el 6 julio de 2021: Dra. Isabel Chirivella y Dra. Ana Beatriz Sánchez-Heras
1.- Introducción:
Los cánceres son enfermedades debidas a la multiplicación de manera autónoma y sin control a partir de una célula dando lugar a muchas nuevas células que invaden los tejidos localmente y a distancia. Se puede deber a agentes externos, químicos (como los componentes del tabaco), físicos (fibras de asbesto, radiaciones ionizantes) y biológicos (como el virus del papiloma, bacterias) o a fallos en los procesos celulares endógenos que alteran o dañan el ciclo celular. Se conocen más de 200 tipos diferentes de cánceres, siendo los más frecuentes los cánceres de piel, el cáncer de pulmón, el cáncer de mama y el cáncer de colon.
Asesoramiento Genético en Cáncer
1.- ¿El cáncer se puede heredar?
Se estima que el 70-80% de los tumores son esporádicos. Hasta el 15-20% de los pacientes con cáncer refieren antecedentes familiares previos, lo que se denomina agregación familiar. Puede ser debido a factores ambientales comunes, estilos de vida similares, o a factores genéticos. En el 5-10% de los casos se puede identificar una alteración en un gen concreto que explica el desarrollo de los cánceres en los miembros de la familia (Figura 1).
La alteración genética, mutación o variante patogénica, suele ocurrir en un gameto (óvulo o espermatozoide). A partir de la fecundación, dicha mutación está en todas las células del nuevo ser, que a su vez la puede transmitir a su descendencia. La mayoría de los síndromes de predisposición a cáncer hereditarios siguen un patrón de herencia autosómica dominante, con una probabilidad de heredar la mutación familiar del 50%. Solo cuando en una célula se produce mutación o pérdida de heterocigosidad del segundo alelo del gen, la función de la proteína se pierde y se desencadena el proceso hacia la malignización.
Es importante destacar que ser portador de una mutación no es sinónimo de padecer cáncer, significa tener un mayor riesgo o probabilidad de desarrollar uno o varios tipos de determinados cánceres.
2.- ¿Cuándo debemos sospechar una predisposición hereditaria al cáncer?
Cuando ha habido varios casos del mismo tipo de cáncer o cánceres relacionados (varía según cada síndrome) en familiares de primer grado (padres, hijos, hermanos) de generaciones sucesivas. Cuando se diagnostica a edad joven (antes de los 40-50 años de edad) un cáncer relacionado con el envejecimiento (cáncer de mama, cáncer de colon, cáncer de próstata). Cuando un individuo presenta tumores bilaterales, multifocales, o más de un tipo de tumor. Y en algunos tumores concretos como el cáncer de ovario de tipo seroso de alto grado, o el carcinoma de los plexos coroideos, por ejemplo.
3.- Consejo genético:
La baja incidencia y prevalencia de los síndromes de predisposición a cáncer hereditario, la complejidad de las diferentes etapas del proceso de consejo genético y el abordaje multidisciplinar hace necesaria la creación de Unidades de Consejo Genético en Cáncer o de Cáncer Familiar y Hereditario.
Las Unidades de Consejo Genético en Cáncer, o también denominadas Consulta de Cáncer Familiar y Hereditario, son áreas de trabajo que dedican su labor asistencial e investigadora al cáncer hereditario y al asesoramiento genético. El consejo genético se puede definir como el proceso de comunicación que se ocupa de los problemas humanos asociados con la aparición, o riesgo de aparición, de una enfermedad genética en una familia. El consejo genético no es tan sólo un test de laboratorio.
Se ha definido el consejo genético como proceso y no como un acto médico aislado. Durante el asesoramiento genético se evalúa el riesgo personal y familiar de susceptibilidad hereditaria al cáncer, la posibilidad de realizar un diagnóstico molecular, mejorar la estimación del riesgo, adecuar las estrategias de seguimiento y de reducción del riesgo. Hay de destacar que no se valoran únicamente aspectos individuales de la persona que acude a la consulta, también la información familiar.
Estas consultas deben coordinarse con los dos niveles asistenciales: Atención Primaria y Atención Especializada. Así mismo, debe existir una relación fluida entre el clínico y el laboratorio que hace el diagnóstico genético molecular.
El proceso del consejo genético se divide en 4 etapas:
3.1.- Identificación de individuos o familias a riesgo:
En la 1ª consulta se procede a evaluar si nos encontramos realmente ante un proceso de cáncer hereditario o familiar. Para ello, debemos recoger los datos de filiación, aspectos de interés sobre otros factores exógenos de riesgo oncológicos (hábitos de salud, hábitos tóxicos, etc.) e interrogaremos de manera detallada el pedigrí o historia familiar de la persona que consulta. En él recogeremos todos los datos referentes a la historia oncológica de la familia: tipos de cáncer, quién los ha padecido, a qué edad, y si ha fallecido, a qué edad y cuál fue la causa. De igual manera recogeremos datos de otras patologías que puedan relacionarse con síndromes hereditarios concretos y que nos puedan ayudad a encuadrar a esa persona en un síndrome exacto.
Tras la realización de esta historia se puede, en algunos casos, informar a la persona consultante sobre la posibilidad de que haya un síndrome hereditario o no y también de las posibilidades de desarrollar cáncer. Se dispone de programas y aplicaciones informáticas, así como de tablas de riesgo que ayudan en el asesoramiento.
Si se identifica un síndrome hereditario concreto se explican los aspectos clínicos, moleculares y de manejo médico para que, con esta información, comprenda las posibilidades que la asistencia sanitaria ofrece para ayudarle a convivir con esa enfermedad, la posibilidad de padecerla, así como orientarle sobre la necesidad o no de realizar un test genético.
Aunque se conozca el gen alterado que produce la enfermedad no siempre se pueden ofrecer medidas preventivas eficaces..
3.2.- Consejo genético pre·test y la firma del consentimiento informado:
En la 2ª consulta, y tras un tiempo de reflexión para decidir si realiza o no el test genético, se procede a la extracción de sangre para el estudio de los genes que puedan estar implicados en el síndrome que se sospecha. Es indispensable que firme un consentimiento informado autorizando la realización del mismo.
En la etapa de consejo genético pre·test se proporciona información sobre los riesgos, beneficios y limitaciones del estudio genético.
Los riesgos del estudio genético son: trastornos psicológicos (ansiedad, culpabilidad, autoestima o depresión), perdida de privacidad, cambios en la dinámica familiar y una falsa sensación de seguridad.
Los beneficios del estudio genético son que explican la aparición de tumores en la familia, permitiendo individualizar el riesgo de cada individuo. De esta manera se ofrecerán medidas de prevención o de diagnóstico precoz y puede ayudar a plantear opciones reproductivas para evitar la transmisión de una mutación.
Las limitaciones del estudio genético son: la posibilidad de un resultado no concluyente o variantes de significado desconocido; los resultados indican probabilidad no certeza de desarrollar cáncer y que no siempre existen medidas de intervención eficaces.
3.3.- Realización del test genético:
En la 3ª consulta se procederá a explicar al probando el resultado del test. Generalmente se realiza un rápido recordatorio del síndrome hereditario ante el que nos encontramos y las implicaciones que va a tener el conocer el resultado del test tanto para él como para el resto de sus familiares.
Con la idea clara sobre lo que significa ese resultado, empezaremos a comentar las opciones de manejo a la hora de intentar reducir el riesgo o detectar de manera precoz el posible cáncer. Lo habitual es que en esa consulta se proceda a tratar temas muy preliminares sobre estos aspectos, de manera que en la próxima visita se plantee el programa definitivo de manejo.
En esta visita también se informará sobre la posibilidad de que otras personas de su familia se encuentren en riesgo y que sería conveniente que contactase con ellas para que inicien el proceso de asesoramiento, si así lo consideran oportuno.
Los resultados del test genético se pueden clasificar en 2 grandes grupos:
3.3.1.- Test informativo:
Es aquel resultado que nos permite asesorar correctamente a la persona que se ha realizado dicho test. Dentro de este grupo habrían 2 opciones:
3.3.1.1.- Verdadero positivo:
Es aquella situación en la que, conociendo previamente el estatus de portador de la familia para una determinada mutación patogénica, detectamos a un portador sano de la misma mutación. En este caso podremos saber de manera más concreta ante qué riesgo de cáncer nos encontramos, así como las opciones que hoy en día se plantean para ese grupo poblacional.
3.3.1.2.- Verdadero negativo:
Es aquella situación en la que, conociendo previamente el estatus de portador de la familia de una determinada mutación patogénica, al realizar el estudio en una persona sana de la familia, no se detecta la mutación. En este caso podemos informar a esa persona de que el riesgo de padecer alguno de los tumores asociados a ese síndrome es similar al de la población general y que no debe someterse a ningún tipo de seguimiento especial, salvo el recomendado a la población general.
3.3.2.- Test no informativo:
Es aquel resultado negativo en ausencia de mutación conocida en una familia con criterios clínicos de alto riesgo para un síndrome hereditario concreto.
Conocer si una persona es portadora de una mutación germinal en un gen de predisposición a padecer cáncer es esencial en el manejo del probando y de su familia. Sin embargo, en los síndrome más prevalentes y en los cuales conocemos los genes implicados en el desarrollo de estas enfermedad, no podemos detectar la alteración que buscamos en la totalidad de los casos. Pero, ¿por qué ocurre esto? Existen distintas razones por las que no somos capaces de detectar mutaciones en todas aquellas familias etiquetadas como de alto riesgo:
3.3.2.1.- Problemas con las técnicas de análisis molecular:
Ninguna técnica es perfecta. Por lo que, en ocasiones, podremos no detectar la alteración aunque se halle presente.
3.3.2.2.- Selección equívoca del probando:
Es esencial que seleccionemos a la persona con más posibilidades de ser portadora de la mutación. Para ello intentaremos que el test se le realice a aquella persona que ha padecido cáncer, que el tipo de cáncer sea uno de los que definen el síndrome, y a ser posible, que lo haya padecido a edad joven.
3.3.2.3.- Fenocopia:
Es habitual que se piense que todos los cánceres que existen en una familia con predisposición son debidos a mutaciones germinales. Sin embargo, en una familia con un síndrome hereditario de cáncer pueden existir casos de cáncer esporádico que nos llevan a error a la hora de interpretar el resultado.
3.3.2.4.- Implicación de genes desconocidos:
Lamentablemente, nuestros conocimientos actuales sobre los genes que están implicados en los síndromes hereditarios no llegan, ni de lejos, a explicar molecularmente por qué se producen esos tumores. Es lógico pensar que deben existir otros genes que aún no se han descubierto que expliquen por qué en una determinada familia claramente hereditaria no hayamos encontrado ninguna mutación.
3.3.2.5.- Resultado de significado desconocido:
Cuando se identifica una mutación de la cual desconocemos su significado patogénico y por tanto no tiene utilidad ni aplicabilidad clínica.
3.4.- Consejo genético post·test y el seguimiento
En esta visita se asientan las recomendaciones definitivas sobre las medidas a tomar y se evalúa el impacto psicológico que el resultado del test ha producido. La evaluación psicológica se va realizando a lo largo de todo el proceso, pero en esta visita tiene una especial repercusión debido a que la persona ha tenido un tiempo para procesar toda la información y las posibilidades de manejo.
4.- ¿Quién se puede beneficiar de un test genético?
Toda persona que sepa que en su familia existen varios casos de cáncer debería acudir a su médico de atención primaria para una primera valoración. Si su médico considera que hay datos que sugieren un trastorno hereditario deberá remitirle a la unidad de consejo genético más cercana (ver mapa de las unidades de cáncer hereditario y familiar de España).
5.- ¿Cuáles son los síndromes hereditarios más frecuentes?
Los principales síndromes de cáncer hereditario son: cáncer de mama y ovario hereditario, cáncer de colon no polipósico o síndrome de Lynch y poliposis adenomatosa familiar (tabla 1).
Figura 1. Eiotlogía del Cáncer.

Tabla 1: Principales síndromes de predisposición a cánceres hereditarios

Poliposis Adenomatosa Familiar
1.- Introducción:
La poliposis adenomatosa familiar (PAF) tiene una incidencia estimada en la población española de 2,8 por cada 100000 habitantes. Se debe a variantes patogénicas en el gen APC. Es causante de menos de un 1% de los cánceres de colon y recto (CCR), pero es importante porque aproximadamente el 100% de los pacientes que no reciben un tratamiento adecuado desarrollan CCR.
Si existen más de 100 pólipos se denomina PAF clásica y si el número de pólipos es entre 20 y 100 pólipos hablamos de PAF atenuada.
Entre un 11 y un 25% de los pacientes no presentan historia familiar de la enfermedad, debido a mutaciones de nueva aparición o “de novo”.
Alteraciones en otros genes producen poliposis adenomatosa de colon y CCR, como MUTYH, POLE, POLD1, AXIN, NTHL1, MSH3 o GREM1. Habitualmente son formas atenuadas, por lo que el riesgo de CCR es menor que en la PAF asociada a APC. .
2.- Gen APC:
El gen APC es un gen supresor de tumores, localizado en el cromosoma 5q. A nivel celular se debe producir una segunda mutación en el otro alelo para que exista una pérdida completa de la función del gen, y dar lugar a la proliferación epitelial y desarrollo de un CCR.
3.- Historia natural:
La PAF se caracteriza por un número creciente de pólipos adenomatosos en el intestino grueso, principalmente en recto y sigma, y en menor medida en el resto del tracto gastrointestinal. El número varía entre cientos y millares. Inicialmente son benignos, pero tienen una tendencia elevada a la degeneración de forma que antes de los cincuenta años la mayoría de los pacientes desarrollarán uno o varios CCR.
Durante años la clínica es silente, con deposiciones más blandas, a veces diarreicas, y ocasionalmente dolores cólicos tipo retortijón. Cuando aparece rectorragia un 30% de los pacientes tienen ya degeneración en uno a más pólipos. La edad media de diagnóstico de CCR es de 39 años y la muerte suele acontecer a los 40-45 años, unos 20 años antes de la edad por fallecimiento por CCR esporádico.
4.- Manifestaciones extracolónicas:
La PAF puede llevar asociada alteraciones en otros órganos como osteomas, anomalías dentarias, pólipos gástricos, pólipos duodenales, quistes epidermoides, hipertrofia congénita del epitelio pigmentario de la retina, tumores desmoides y tumores malignos extracolónicas como hepatoblastoma, cáncer de tiroides, del árbol biliar, de páncreas y tumores del sistema nervioso central (tabla 1).
5.- Seguimiento clínico:
La evaluación clínica basal incluye el estudio de fondo de ojo y una ortopantomografía basal.
El cribado del CCR se realiza mediante sigmoidoscopia flexible bianual, que se inicia entre los 12-15 años en la FAP clásica y a los 18-20 años en las formas atenuadas. En el momento en que se detecta algún adenoma se realiza seguimiento mediante colonoscopia total.
El seguimiento de las manifestaciones del tracto gastrointestinal alto se realiza mediante endoscopia digestiva alta, a partir de los 25-30 años. Para los pólipos duodenales se sigue una periodicidad según la clasificación por estadios de Spigelman: estadio 0 cada 4 años, estadio I cada 2-3 años, estadio II cada 1-3 años, estadio III cada 6-12 meses. En caso de estadio IV se recomienda cirugía.
Así mismo, se recomienda exploración clínica anual con palpación y ecografía tiroidea, que se inicia a partir de los 25-30 años de edad.
En niños se puede realizar cribado del hepatoblatoma mediante determinación sérica del marcador tumoral αFP, palpación y ecografía abdominal cada 3-6 meses, hasta los 5 años de edad.
6. Manejo clínico de la poliposis adenomatosa familiar:
6.1.- Manejo clínico de la afectación colónica en la poliposis adenomatosa familiar:
La afectación colónica de la PAF debe tratarse mediante colectomía profiláctica. En general, se acepta que la colectomía puede realizarse con seguridad una vez transcurrida la pubertad y sólo debe realizarse cuando no se puede garantizar un adecuado control endoscópico de los pólipos, por presentar un número importante de pólipos mayores de 5 milímetros o adenomas de alto grado de displasia.
6.2.- Manejo de la afectación duodenal:
El tratamiento de los pólipos gastroduodenales varía según su localización. Lo pólipos fúndicos, una vez confirmados su carácter hiperplásico no necesitan tratamiento. En el duodeno la mejor opción en los pólipos aislados es la polipectomía endoscópica. Cuando la afectación duodenal es grave el tratamiento recomendado es la duodenopancreatectomía cefálica con preservación de píloro y anastomosis pancreatogástrica.
7.- Quimioprevención:

Síndrome de Lynch
1.- Introducción:
Tan sólo un 5-6% de los CCR se consideran hereditarios. El síndrome de Lynch o de Cáncer de Colon Hereditario No Polipósico es el principal responsable. Es debido a mutaciones en los genes MLH1, MSH2, MSH6 y PMS2 que codifican para proteínas que intervienen en el sistema de reparación del ADN mismacht repair (MMR) y a deleciones del gen EPCAM que producen silenciamiento de MSH2.
En el síndrome de Lynch el CCR se caracteriza por aparecer a una edad media más temprana que los casos esporádicos, localizarse más frecuentemente en el colon derecho y con frecuencia aparecen tumores sincrónicos o metacrónicos. El segundo tumor más frecuente es el cáncer de endometrio, seguido de cáncer de ovario y cáncer de estómago.
Aproximadamente en un 50 - 60% de las familias con criterios clínicos de Síndrome de Lynch no se identifica una mutación en los genes MMR. En la actualidad, el uso de paneles de genes múltiples por secuenciación masiva ha permitido detectar variantes patogénicas otros genes como POLE, POLD1, MUTYH, APC, BRCA1, BRCA2, CHEK2 o ATM. Hay familias con agregación de casos de CCR exclusivamente, denominado Carcinoma Colorrectal Familiar tipo X, en los que todavía no se ha podido identificar un gen o genes responsables.
2.- Criterios para remitir a la unidad de consejo genético en cáncer:
Hay unos criterios clínicos de sospecha, Criterios de Ámsterdam y Criterios de Bethesda, así como modelos matemáticos que calculan la probabilidad de encontrar una mutación en los genes asociados a este síndrome, como MMRpredict, MMRpro, y PREMM5, pero el análisis del tumor es el método diagnóstico fundamental.
Se deben derivar a todos los pacientes o familias que cumplan todos los criterios de Amsterdam o alguno de los criterios de Bethesda modificados: un carcinoma colorrectal diagnosticado antes de los 50 años, un cáncer de colon sincrónico o metacrónico, un caso de cáncer colorrectal y la presencia de otro tumor relacionado con el síndrome de Lynch, un cáncer colorrectal y 1 familiar de primer grado con un tumor asociado al síndrome de Lynch antes de los 50 años, un cáncer colorrectal y 2 familiares de primer o segundo grado con un tumor asociado al síndrome de Lynch (independientemente de la edad). Sin embargo, la sensibilidad es baja.
Por ello actualmente se reconoce que es coste-eficaz hacer cribado realizando inmunohistoquimia de las de proteínas reparadoras MMR en todos los CCR diagnosticados antes de los 70 años y en todos los cánceres de endometrio. La identificación de pérdida de expresión de alguna de las proteínas MLH1, MSH2, MSH6, y PMS2 es criterio de remisión a una Unidad de Consejo Genético en Cáncer, y permite dirigir el estudio genético de diagnóstico de S. Lynch.
3.- Riesgo de otros tumores en portadores de mutaciones CCHNP:
Las personas con síndrome de Lynch presentan un mayor riesgo de desarrollar diferentes tipos de cánceres: CCR (24-75%), endometrio (27-71%), ovario (3-13%), estómago (2-13%), vías urinarias (1-12%), intestino delgado (4-7%), vías biliares (2%), y sistema nervioso central (1-4%).
4.- Medidas de reducción de riesgo tras la detección de mutación en CCNPH:
4.1- Seguimiento:
4.1.1.- Seguimiento para la detección precoz de carcinoma de colon:
La colonoscopia ha demostrado disminuir la incidencia y la mortalidad por CCR en pacientes afectos de S. Lynch.
El riesgo de desarrollar un CCR antes de los 25 años es bajo. Por ello, se recomienda realizar una colonoscopia cada 1 - 2 años, iniciándola a los 25 años y realizar una colonoscopia anual a partir de los 40 años.
4.1.2.- Seguimiento para la detección precoz del carcinoma de endometrio:
Se recomienda exploración ginecológica y ecografía transvaginal con aspirado endometrial anual, a partir de los 30-35 años de edad.
4.1.3.- Seguimiento para la detección precoz de neoplasias extracolónicas:
El diagnóstico precoz de cáncer gástrico tan sólo se recomienda en países de alta incidencia como países del lejano oriente, o si hay agregación familiar. Se recomienda endoscopia digestiva alta anual desde los 30-35 años.
El cribado de tumores urológicos y de ovario se recomiendan si en la familia hay algún caso.
Para el cáncer de ovario se recomienda una exploración ginecológica, ecografía transvaginal y la determinación del marcador tumoral serológico CA 125.
Para carcinomas urológicos citologías de orina y ecografía urológica cada 1-2 años, desde los 25-30 años.
No hay la suficiente evidencia para realizar el cribado de otros tumores como el cáncer de páncreas, tracto biliar, intestino delgado, tumores SNC, etc.
4.2.- Quimioprevención:
Los resultados del ensayo clínico CAPP2, con aspirina a dosis de 500 mg oral diariamente durante al menos dos años, muestran disminución de CCR y de otros tumores, siendo el beneficio clínico evidente después de los 5 años.
4.3.- Cirugía reductora de riesgo:
4.3.1.- Cirugía reductora de riesgo de carcinoma de colon:
No disponemos de evidencia para recomendar una colectomía profiláctica en las personas diagnosticadas de síndrome de Lynch.
4.3.2.- Cirugía reductora de riesgo de carcinoma de endometrio y de ovario:
La histerectomía más doble anexectomía elimina el riesgo de desarrollar tumores ginecológicos, frente al 33% de desarrollar un carcinoma de endometrio y un 5,5% de ovario.
4.4.- Seguimiento del carcinoma colorrectal familiar tipo X:
En familias sin evidencia de un defecto en los genes MMR se recomienda realizar un seguimiento menos intensivo con colonoscopia cada 3 años desde los 45 años o 10 años antes del primer diagnostico en la familiar.
No precisa cribado de carcinoma de endometrio, puesto que no presentan riesgo de desarrollar tumores asociados al síndrome de Lynch.
Tabla 1: Seguimiento en personas con diagnóstico de síndrome de Lynch

Cáncer de Mama y Ovario Hereditario
1.- Introducción:
Aproximadamente un 7% de los cánceres de mama y un 11-15% de los cánceres de ovario se consideran hereditarios, debido a una mutación genética heredada de uno de los padres.
BRCA1 y BRCA2 son los genes que se asocian con una mayor proporción de casos al cáncer de mama y ovario hereditario, en concreto el cáncer de ovario seroso de alto grado. Se han descrito otros genes de alta penetrancia asociados al cáncer de mama: TP53 (síndrome de Li-Fraumeni), PTEN (síndrome de Cowden), STK11 (síndrome de Peutz-Jeghers) o PALB2; otros genes de moderada penetrancia como ATM, CHEK2 y NBN. Y otros genes asociados al cáncer de ovario: genes MMR, RAD51C, RAD51D y BRIP1.
2.- Genes de alta penetrancia BRCA1/BRCA2 y PALB2:
El gen BRCA1 está localizado en el brazo largo del cromosoma 17 (17q21) y el gen BRCA2 en el cromosoma 13 (13q12). Las proteínas BRCA1 y BRCA2 actúan en las vías de reparación del ADN y su inactivación mediante mutación provoca indirectamente la aparición del tumor por acumulación de mutaciones en otros genes reguladores directos del ciclo celular.
El gen PALB2 (partner and localizer of BRCA2) es un gen relacionado con la anemia de Fanconi. Codifica una proteína que posee un papel esencial en el mantenimiento del genoma, reparando la rotura de la doble cadena de ADN. La función de la proteína PALB2 se desarrolla conjuntamente con BRCA1 y BRCA2 en la misma vía de reparación del ADN.
3. Genes de moderada penetrancia que aumentan el riesgo de cáncer de mama
Los siguientes genes de moderada penetrancia aumentan el riesgo de cáncer de mama en menor medida.
3.1. Gen ATM
ATM o gen ataxia telangiectasia mutado, es un gen localizado en el brazo largo del cromosoma 11, codifica la proteína ATM serina/treonina quinasa que está implicada en la regulación de los procesos de control de la división celular y en la reparación de daños sufridos por la molécula de ADN.
El gen ATM puede aumentar el riesgo de cáncer de mama. En un meta-análisis se estimó un riesgo relativo de 2.8 (90% IC, 2.2-3.7, p>0.001). Otros análisis muestran un 1% de mutaciones en mujeres con cáncer de mama. Y un estudio holandés de mujeres con cáncer de mama a edad temprana detectó un 8.5% de mutaciones del gen ATM.
3.2 Gen CHEK2
El gen CHEK2 (cell cycle checkpoint kinase 2) es otro de los genes identificados que aumenta el riesgo de cáncer de mama. Regula los puntos de control del ciclo celular y la apoptosis en respuesta al daño en el ADN, particularmente roturas del ADN de doble cadena.
En un estudio que no detectó mutaciones en BRCA1/2 en mujeres con una fuerte historia familiar de cáncer de mama/ovario se detectó en el 5% de estas. El riesgo acumulado de cáncer de mama en mujeres con mutación de CHEK2 e historia familiar de cáncer de mama se ha estimado entre el 28 y el 37%, siendo mayor en mujeres con antecedentes de cáncer de mama familiar. El riesgo estimado es de 3.0 (90% IC, 2.6-3.5).
Se ha estudiado la asociación de la variante truncada 1100delC con el riesgo de cáncer de mama en mujeres no seleccionadas por su historia familiar, detectando un riesgo relativo de 2.34 en el estudio del consorcio europeo y australiano. Sin embargo, esta mutación no se ha comprobado que esté presente en la población española de cáncer de mama familiar.
3.3. Gen NBN
El gen NBN es responsable de sintetizar la proteína nibrina. La nibrina es una proteína asociada con la reparación de roturas de doble hélice del ADN que ocasionarían serios daños al genoma.
Las mujeres con mutaciones heterocigotas en este gen tienen un aumento del riesgo de cáncer de mama con un RR 3.1. Un meta-análisis incluyendo 7 estudios mostró un aumento significativo con la variante 657del5 (RR 2.42).
4. Genes de alta y moderada penetrancia que aumentan el riesgo de cáncer de ovario
Hay otros genes de la familia de la proteína RAD51, involucrados en la recombinación homóloga y reparación del ADN como RAD51C y RAD51D, o el gen BRIP1, que aumentan el riesgo de CO, pero no hay evidencia clara sobre el aumento del riesgo de CM.
En la población general, el riesgo de CO, trompa de Falopio o primario peritoneal es del 1,5%. Las mutaciones en los genes BRCA1/2, RAD51C, RAD51D y BRIP1 incrementan más de 5 veces el riesgo de desarrollar CO en mujeres portadoras de alteraciones en estos genes en comparación con la población general.
4.1. RAD51C y RAD51D
RAD51 es una proteína de 339 aminoácidos que desempeña un papel importante en la recombinación homóloga del ADN. La familia de genes RAD51, que incluye los genes RAD51C y RAD51D, está involucrada en la reparación del ADN mediante la interacción con los genes BRCA1 and BRCA2. En diferentes estudios, se ha concluido que RAD51C es un gen de predisposición al cáncer de ovario, y sus mutaciones se pueden encontrar en el 1% de pacientes afectas de CO. En mujeres con mutaciones en RAD51C el riesgo de CO es del 11% a los 80 años. En mujeres portadoras de mutación en el gen RAD51D el riesgo de CO a los 80 años es de un 13%.
4.2. BRIP1
BRIP1 es una proteína del grupo de anemia de Fanconi cuya función es la reparación de ADN de doble cadena. Esta proteína también parece ser importante en el CO, donde parece actuar como supresor de tumores. Las mutaciones en BRIP1 se asocian con un riesgo de hasta el 5.8% a los 80 años de CO.
5. Criterios para remitir a la unidad de consejo genético en cáncer:
La complejidad y extrema laboriosidad del estudio de ambos genes y la escasa prevalencia de mutaciones en la población hacen inviables los análisis poblacionales.
Los criterios clínicos para indicar un estudio de los genes relacionados con el cáncer de mama/ovario hereditario están basados en la historia personal y familiar (se pueden consultar en “SEOM clinical guidelines in hereditary breast and ovarian cancer (2019)”. PMID:31889241. DOI:10.1007/s12094-019-02262-0)
6. Riesgo de cáncer en mutaciones BRCA1/BRCA2:
6.1. Riesgo de cáncer de mama y ovario en portadores de mutaciones BRCA1/BRCA2
La penetrancia, definida como la probabilidad de que un portador de una mutación en BRCA1/2 desarrolle un cáncer a lo largo de su vida, suele expresarse como el riesgo acumulado de cáncer a los 70 años. La penetrancia deriva del grado de agregación familiar de cáncer.
En un meta-análisis realizado sobre la penetrancia de BRCA1 y BRCA2, a partir de los principales estudios publicados, se recoge que el riesgo acumulado de cáncer de mama a los 70 años es del 57% (95% IC, 47 a 66%) para portadores de mutación en BRCA1 y del 49% (95% IC, 40 a 57%) en BRCA2; en cuanto al cáncer de ovario seroso de alto grado se estima un riesgo acumulado a los 70 años del 40% (95% IC, 35 a 46%) para portadores de mutación en BRCA1 y del 18% (95% IC, 13 a 23%) en BRCA2.
En un estudio publicado en 2017 con una cohorte prospectiva de 6036 mujeres portadoras de mutación en el gen BRCA1 y 3820 en el gen BRCA2, el riesgo en portadoras de mutación BRCA1 de CM fue del 78% (95% IC 65% a 79%) y de CO del 44% (95% IC 36% a 53%), mientras que en portadoras de mutación en BRCA2 el riesgo de CM fue del 69% (IC 95% 61 a 77%) y de CO del 17% (95% IC 11 a 25%). La incidencia aumenta desde los 30 a 40 años en las portadoras de mutación en BRCA1 y de los 40 a 50 años para BRCA2, y se mantiene elevado a lo largo de toda la vida. El carcinoma ductal in situ de mama (CDIS) también debe considerarse como uno de los tumores que se asocian a mutaciones en estos genes. Aparece a una edad más temprana que en la población general, unos 12 años antes.
Respecto al riesgo de CM en los hombres es mayor en los portadores de mutación en BRCA1/2 respecto a la población general. La probabilidad de desarrollar CM en portadores de mutación en BRCA2 es del 8%, y del 1% en los portadores de mutación en BRCA1. En la población general el riesgo de CM es del 0.1%.
6.2. Riesgo de otros tumores en portadores de mutación BRCA1/2
Los portadores de mutaciones en BRCA1 presentan además de un riesgo elevado de CM y de CO riesgo de otros tumores. En el estudio del Breast Cancer Linkage Consortium (2002), los portadores de mutación BRCA1 presentaban un aumento del riesgo de cáncer de páncreas (RR=2,26), carcinoma de endometrio (RR=2,65), cáncer de cérvix (RR=3,72), y de cáncer de próstata en menores de 65 años (RR=1,82) (el riesgo de cáncer de próstata en mayores de 65 años no estaba aumentado).
Respecto a los portadores de mutaciones en BRCA2 también se ha documentado un aumento del riesgo de otros tipos de tumores, como cáncer de próstata. En uno de los estudios con más mujeres realizado del Breast Cancer Linkage Consortium (3728 individuos con mutación, 681 de ellos con cáncer de mama/ovario, entre 173 familias con cáncer de mama/ovario y mutación en BRCA2), estudiaron la frecuencia de otros cánceres en los familiares de 1er grado, encontrando un aumento del riesgo estadísticamente significativo en el cáncer de próstata (RR=4,65), cáncer de páncreas (RR=3,51), cáncer de vesícula biliar y conductos biliares (RR=4,97), estómago (RR=2,59), y melanoma maligno (RR=2,58); el riesgo de cáncer de próstata en hombres menores de 65 años es de 7,3. Por último, mutaciones en este gen aumentan la predisposición al cáncer de mama en varones. Es frecuente que las familias de alto riesgo con casos de cáncer de mama en varones posean mutaciones en BRCA2.
Con los datos de estudios actuales podemos resumir el riesgo de tumores asociados a BRCA1/2:
Riesgo de tumores ginecológicos:
- Cáncer en las trompas de Falopio. Un 50% de los tumores serosos en portadoras de mutaciones BRCA1/2 tienen un origen distal a las trompas de Falopio. El riesgo en la población general es del 0.2%. En un estudio retrospectivo de 108 mujeres con carcinoma en las trompas, un 33% eran portadoras de mutación BRCA1/2. Además, en mujeres portadoras es más frecuente antes de los 60 años vs no portadoras (40.3 vs 17.4%).
- Tumor primario peritoneal (es un tumor maligno poco frecuente de la cavidad peritoneal de origen extra-ovárico, con clínica e histología similar a los estadios avanzados del carcinoma seroso de alto grado de ovario). La probabilidad de desarrollo en portadoras de mutación es del 1.3%.
- Tumores de endometrio. No parece que esté aumentado en mujeres portadoras de mutación en los genes BRCA1/2. El aumento del riesgo parece estar relacionado con el tratamiento con tamoxifeno.
Cáncer de páncreas
Como se ha comentado previamente el riesgo de cáncer de páncreas es mayor en portadores de mutaciones BRCA, entre el 1 y 4.9% para BRCA1 y BRCA2 respectivamente. La mediana de edad en portadores de mutación en BRCA2 es de 67 años en los hombres y de 59 años en las mujeres.
Cáncer de próstata
En hombres portadores de mutación en BRCA2, el riesgo de cáncer de próstata es entre 5 y 9 veces el de la población general a los 65 años, lo que significa un riesgo del 33%. También se ha descrito que aparece de forma más precoz y con una mayor agresividad.
Riesgo de otros tumores
Cáncer de colon. Los datos sobre el aumento del riesgo de cáncer de colon en estos pacientes no son consistentes. Algunos estudios describen el aumento del riesgo en BRCA1, pero otros estudios no confirman esta asociación.
Melanoma. Se ha descrito asociación entre mutaciones en BRCA2 y riesgo de melanoma, pero este riesgo no está bien caracterizado. No se ha visto una clara asociación a mutaciones en BRCA1.
Tumores de estómago y vías biliares. Parece que hay aumento del riesgo de estos tumores, pero no ha sido cuantificado.
7. Riesgo de cáncer en mutaciones PALB
Las mutaciones en este gen aumentan el riesgo de cáncer de mama. Un meta-análisis de 3 estudios estimó un riesgo relativo (RR) de 5.3 (90% IC, 3.0-9.4). El riesgo de cáncer de mama asociado a mutaciones en PALB2 aumenta con la edad, con un riesgo del 13-21% a los 50 años y del 44-63% a los 80 años de edad. Este riesgo aumenta con el número de familiares afectos, de modo que el riesgo de cáncer de mama a los 70 años si no hay familiares con cáncer de mama es del 33% comparado con el 58% en las que tienen al menos 2 familiares con cáncer de mama.
El riesgo de cáncer de ovario a lo largo de la vida en portadoras de mutación en PALB2 es del 3-5% a lo largo de la vida. Las medidas de seguimiento se deberán adoptar según la historia familiar.
8. Medidas de reducción de riesgo tras la detección de mutación en BRCA
Aunque no existe un nivel de evidencia alto, se recomienda a las mujeres con alto riesgo de cáncer de mama reducir la ingesta calórica total, evitar la obesidad, realizar ejercicio físico con regularidad y moderar el consumo de alcohol.
8.1.- Seguimiento:
8.1.1.- Cribado del cáncer de mama:
La vigilancia del cáncer de mama incluye la autoexploración mamaria mensual indicada en mujeres mayores de 18 años, la exploración mamaria clínica cada 6 meses a partir de los 25 años, la mamografía anual con o sin ecografía mamaria en mujeres mayores de 30 años y hasta los 75 años y la resonancia magnética mamaria anual en mujeres mayores de 25 años y hasta los 70 años.
8.1.2.- Cribado de cáncer de ovario:
En mujeres portadoras de mutación BRCA se recomienda realizar una exploración ginecológica con ecografía transvaginal y la determinación sérica del marcador tumoral CA 125, cada 6-12 meses desde los 30 años.
8.1.3.- Seguimiento en varones portadores de la mutación BRCA:
En varones portadores de la mutación BRCA el riesgo de desarrollar carcinoma de mama es menor. Por ello, tan sólo se recomienda la autoexploración mamaria, y en caso de detectarse alguna anomalía se realizará una mamografía con o sin ecografía mamaria.
Se recomienda realizar el cribado de carcinoma de próstata mediante tacto rectal y la determinación sérica del marcador tumoral PSA a partir de los 40 años.
8.1.4.- Cribado de otros tumores:
En mujeres y hombres portadores de mutación en BRCA1 o BRCA2 se indicará el cribado de otros tumores dependiendo de la historia familiar.
8.2.- Quimioprevención:
8.2.1.- Quimioprevención del cáncer de mama:
En el momento actual no disponemos de evidencia científica para recomendar un tratamiento para reducir el riesgo de cáncer de mama en mujeres portadoras de mutación en BRCA sin enfermedad.
8.2.2.- Quimioprevención del carcinoma de ovario:
Los anticonceptivos orales han demostrado disminuir en un 50% el riesgo de desarrollar cáncer de ovario en mujeres portadoras de la mutación BRCA. Aunque, potencialmente podrían causar un ligero incremento del riesgo de cáncer de mama en portadoras de la mutación BRCA1.
8.3.- Cirugía reductora de riesgo en portadoras de mutaciones BRCA1/2:
Constituye la estrategia más efectiva de que disponemos en la actualidad para disminuir el riesgo de cáncer de mama y de ovario, logrando una reducción de riesgo superior al 90% en las mujeres portadoras de mutaciones de los genes BRCA.
8.3.1.- Cirugía reductora de riesgo de cáncer de mama:
La mastectomía bilateral reduce el riesgo de desarrollar carcinoma de mama en un 90% en mujeres portadoras de mutación en BRCA.
La mastectomía subcutánea realiza una exéresis del tejido glandular, y a diferencia de la mastectomía simple preserva la totalidad de la piel de la mama, incluidos la areola y el pezón. La mastectomía subcutánea deja como mínimo un 5% de parénquima mamario, fundamentalmente en la zona retroareolar y en la prolongación axilar, tejido que es teóricamente susceptible de cancerización.
No existe unanimidad en el modelo quirúrgico a seguir para la prevención de mujeres sanas con alto riesgo de padecer cáncer de mama. Generalmente se realiza una mastectomía bilateral profiláctica con reconstrucción inmediata.
8.3.2- Cirugía reductora de riesgo de cáncer de ovario:
La salpingo-ooforectomía bilateral en mujeres con mutación en BRCA1 entre los 35-40 años y en mujeres con mutación en BRCA2 entre los 40-45 años, que hayan cubierto sus deseos reproductivos, es la alternativa más eficaz, con una reducción del 80% del riesgo de cáncer de ovario.
Sin embargo, queda un 5% de riesgo de carcinoma peritoneal primario.
Tras la ooforectomía deben controlarse los efectos de la menopausia precoz en su repercusión sobre la osteoporosis y el perfil lipídico.
9. Medidas de reducción de riesgo tras la detección de mutación en PALB2
En las pacientes con mutaciones germinales del gen PALB2, se recomienda seguimiento mediante mamografía anual desde los 30 años, ya que a esta edad aumenta el riesgo y considerar RNM mamaria anual.
No hay datos sobre el beneficio de reducir el riesgo mediante mastectomía bilateral, por lo que se debe tener en cuenta en función de los antecedentes familiares.
El riesgo de cáncer de ovario no está claro por lo que no hay suficiente evidencia para recomendar cirugía preventiva.
PALB2 se asocia con la anemia de Fanconi heredada de forma autosómica recesiva por lo que hay que discutir las diferentes opciones reproductivas.
10. Medidas de seguimiento en mujeres con mutaciones en genes de moderada penetrancia que aumentan el riesgo de cáncer de mama.
10.1. Mutaciones en el gen ATM
Se recomienda seguimiento mediante mamografía anual desde los 40 años y considerar RNM mamaria según la historia familiar de cáncer de mama.
No hay datos sobre el beneficio de reducir el riesgo mediante mastectomía bilateral, por lo que se debe tener en cuenta con los antecedentes familiares.
Recordar la asociación a la ataxia-telengiectasia de forma recesiva por lo que hay que discutir las diferentes opciones reproductivas.
10.2. Mutaciones en el gen CHEK2
Se recomienda seguimiento mediante mamografía anual desde los 40 años y considerar RNM mamaria anual según la historia familiar.
No hay datos sobre el beneficio de reducir el riesgo mediante mastectomía bilateral, por lo que se debe tener en cuenta con los antecedentes familiares.
10.3. Mutaciones en el gen NBN
Se recomienda seguimiento mediante mamografía anual desde los 40 años, y considerar RNM mamaria anual.
No hay datos sobre el beneficio de reducir el riesgo mediante mastectomía bilateral, por lo que se debe tener en cuenta con los antecedentes familiares.
11. Medidas de seguimiento y cirugías reductoras de riesgo en mujeres con otras mutaciones que aumentan el riesgo de cáncer de ovario.
11.1. Mutaciones en el gen RAD51C y RAD51D
No existen guías de manejo de riesgo de CO en portadoras de RAD51C ni de RAD51D por ello es muy importante adaptarlas a la historia personal y familiar de CO. Debemos tener en cuenta que el diagnóstico de CO en portadoras de mutación en RAD51C generalmente es en mujeres postmenopáusicas, por lo que se puede considerar realizar la Salpingo-Ooforectomia Bilateral Profiláctica (SOBP) una vez la mujer haya alcanzado la menopausia natural. En el caso del gen RAD51D, la opción de SOBP puede considerarse a partir de los 45-50 años.
11.2. Mutaciones en el gen BRIP1
Es importante reseñar que la edad media al diagnóstico de CO en mujeres portadoras de una mutación en BRIP1 es de 63.8 años y un 93% de los casos se diagnostican a edades mayores de los 50 años. Actualmente, solo se recomiendan controles ginecológicos regulares y SOBP a partir de los 50-55 años.
12. Alternativas en mujeres de alto riesgo de carcinoma de mama hereditario tras un resultado genético no informativo
Se aconseja valorar el riesgo según un modelo probabilístico (ej. CanRisk: (https://canrisk.org). Según las guías de la American Cancer Society (ACS) se debe ofrecer vigilancia con RNM mamaria a aquellas mujeres que presenten un riesgo de cáncer de mama mayor del 20-25%, calculado según los modelos de predicción. Se recomienda mamografía y RNM anual iniciando unos 5 años antes de la edad más precoz del cáncer de mama diagnosticado en la familia.
El seguimiento ginecológico específico no es necesario en familiares sin mutación BRCA y sin antecedentes familiares de carcinoma de ovario.
No se recomienda la cirugía reductora de riesgo de carcinoma de mama.
Tabla 1: Criterios clínico·patológicos de alto riesgo de síndrome de cáncer de mama y ovario hereditario
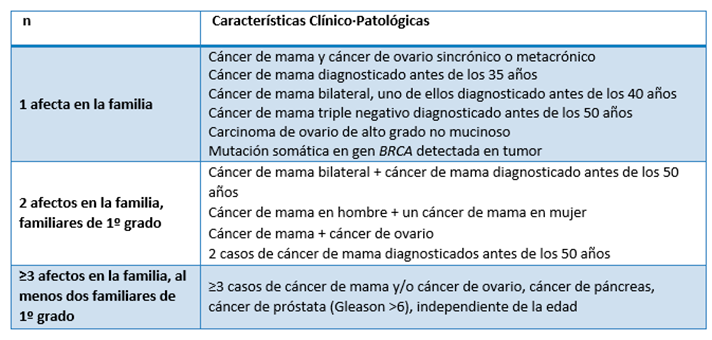
Familiares de primer grado son: madre, hermanas, hijas. No considerar a los hombres al contabilizar grado de parentesco
Cáncer Renal Hereditario
Los carcinomas de células renales representan entre un 2 y un 4% de las neoplasias sólidas malignas. La mayoría de los casos son esporádicos (95%) y se asocia a factores ambientales como tabaco, obesidad, hipertensión arterial, exposición a sustancias químicas o una enfermedad renal con insuficiencia renal terminal.
El carcinoma de células renales hereditario supone un 2-5% de los tumores renales. Se han descrito hasta 10 síndromes que asocian carcinoma de células renales (tabla 1).
Los tumores renales asociados a un síndrome hereditario se caracterizan por una historia familiar de cáncer, se desarrollan a edades tempranas, son bilaterales, multifocales y se asocian a lesiones cutáneas y/o procesos sistémicos.
1.- Síndrome de Von·Hippel·Lindau (VHL)
En Europa el VHL tiene una prevalencia de 1-9 casos por cada 100.000 habitantes.
1.1.- Genética
Se asocia a una mutación en el gen oncosupresor VHL, localizado en el brazo corto del cromosoma 25 (3p25). Presenta una penetrancia casi completa (>90% a los 65 años), con una expresividad variable, incluso dentro de la misma familia.
1.2.- Clínica
La edad media de presentación es de 26 años, aunque puede manifestarse en la infancia. Generalmente debuta con sintomatología oftalmológica o neurológica.
1.2.1.- Hemangioblastomas del Sistema Nervioso Central (SNC)
Los hemangioblastomas del SNC suponen un 1-2% de todos los tumores primarios del SNC. Habitualmente aparecen en el cerebelo, pero también pueden afectar al tronco cerebral o a la médula espinal.
Es la forma de presentación en un 40% de los casos de VHL. Suelen debutar a una edad más joven que los hemangioblastomas del SNC esporádicos: 29 años.
Son tumores benignos, pero pueden provocar clínica por compresión o hemorragia. El comportamiento clínico es impredecible. Pueden estar estables durante años, crecer de forma lineal, exponencial o a saltos.
1.2.1.1.- Manejo de los hemangioblastomas del SNC
La cirugía es el tratamiento estándar, indicada cuando provoca síntomas o la enfermedad presenta un rápido crecimiento. La radioterapia externa o la radiocirugía ofrece resultados inferiores a la cirugía, pero pueden tener un papel interesante en lesiones inaccesibles a la cirugía.
No hay tratamientos médicos aprobados.
1.2.2.- Hemangioblastomas retinianos
Los hemangioblastomas retinianos son la forma de presentación más frecuente. En un 50% de los pacientes son múltiples y bilaterales. Suelen ser asintomáticos, pero pueden causar desprendimiento de retina, edema macular, glaucoma y pérdida de visión.
El tratamiento será conservador, equilibrando el riesgo/beneficio. Cuando los hemangioblastomas retinianos se localizan en la periferia se indica la administración de fotocoagulación por láser de las lesiones de pequeño tamaño y de crioterapia de las lesiones mayores. La radioterapia puede indicarse si fracasan otros métodos.
1.2.3.- Carcinoma de células renales
Aproximadamente 2/3 de los pacientes diagnosticados de VHL presentan quistes renales, con un elevado riesgo de desarrollar carcinoma de células renales (7%). Es la causa más frecuente de muerte en pacientes diagnosticados de VHL.
El cáncer renal es de subtipo células claras, habitualmente multicéntricos y bilaterales. Suele diagnosticarse a una edad media de 44 años, con una penetrancia del 70% a los 60 años de edad. El 85% de los pacientes presentan un segundo tumor a los 5 años y un 30% el tercer tumor a los 10 años. Por el contrario, no parecen tener un alto riesgo de metástasis a distancia.
El manejo de los carcinomas de células renales es conservador: observación en tumores pequeños (<3 cm), nefrectomía parcial, crioterapia o ablación por radiofrecuencia si >3 cm.
1.2.4.- Feocromocitoma
Suelen diagnosticarse en pacientes jóvenes, incluso en edad pediátrica. Con frecuencia son tumores múltiples, extraadrenales (paragangliomas) y asintomáticos. Menos del 7% de los casos pueden ser malignos.
El tratamiento quirúrgico estándar es la adrenalectomía parcial laparoscópica.
1.2.5.- Tumores del saco endolinfático
Los tumores del oído medio corresponden a cistoadenoma papilar del saco endolinfático y están presentes en un 7-14% de los pacientes con VHL. Frecuentemente son bilaterales y suelen localizarse en la porción posterior del hueso petroso temporal. Son tumores de crecimiento lento, que ocasionan destrucción ósea local y provocan clínica súbita o gradual: pérdida auditiva, tinnitus, disfunción vestibular y finalmente parálisis facial.
En pacientes asintomáticos se optará por seguimiento/vigilancia. El tratamiento estándar en los pacientes sintomáticos diagnosticados de cistoadenoma papilar del saco endolinfático es la cirugía. En aquellos pacientes que presenten recidivas locales de la enfermedad se valorará la administración de radioterapia.
1.2.6.- Lesiones pancreáticas
Más de un 70% de los pacientes presentan lesiones en el páncreas. En la mayoría de los casos son quistes (70%), cistoadenomas serosos (9%) y tumores neuroendocrinos (9-17%). No aumenta el riesgo de desarrollar adenocarcinoma de páncreas.
La mayoría de las lesiones son asintomáticas y de lenta evolución. Se recomienda la cirugía en aquellas lesiones >3 cm en cuerpo/cola o >2 cm en cabeza de páncreas. Las lesiones más pequeñas y asintomáticas pueden ser vigiladas.
1.2.7.- Lesiones en el epidídimo
Un 10-60% de los varones diagnosticados de VHL pueden desarrollar quistes y cistoadenomas epididimarios. Los cistoadenomas papilares del epidídimo y ligamento ancho son unilaterales en la población general y bilaterales en VHL, característica casi patognomónica.
No presentan potencial maligno y por lo tanto no requiere la intervención quirúrgica a menos que provoque dolor local.
1.3.- Seguimiento
El seguimiento se centrará en las 3 principales causas de morbi-mortalidad: hemangioblastomas, carcinoma de células renales y feocromocitoma (tabla 2)..
2.- Síndrome de Birt-Hogg-Dubé (BHD)
2.1.- Genética
BHD está causado por mutaciones en el gen foliculina (FLNC), localizado en el cromosoma 17 (17p11.2). Presenta una herencia autosómica dominante, con una penetrancia elevada.
2.2.- Clínica
2.2.1.- Manifestaciones cutáneas
Las manifestaciones cutáneas son fibrofoliculomas, tricodiscomas, acrocordomas, fibromas perifoliculares y angiofibromas.
El tratamiento de las manifestaciones cutáneas se indicará por razones estéticas. El tratamiento de los fibrofoliculomas y tricodiscomas es difícil porque tienden a ser numerosos y recurrentes. Además del tratamiento quirúrgico se ha descrito el uso de isotretinoína oral, láser·C02, láser·erbium: YAG y rapamicina.
2.2.2.- Manifestaciones pulmonares
Las manifestaciones pulmonares son quistes pulmonares (77-89%) y neumotórax espontáneo (24-38%).
Se recomienda evitar el tabaquismo y los cambios de presión como los viajes en avión y el buceo.
2.2.3.- Manifestaciones renales
Las manifestaciones renales son el carcinoma de células renales (6.5-34%) con una edad media de diagnóstico de 48 años.
El tratamiento del carcinoma de células renales es quirúrgico, mediante cirugía conservadora.
2.2.4.- Otras manifestaciones clínicas
Se han descrito otras manifestaciones como oncocitoma de parótida, pápulas orales, patología de tiroides, cáncer de colon, carcinoma escamoso de cabeza y cuello, linfoma de Hodgkin, cáncer de endometrio, cáncer de próstata, cáncer de mama, adenoma adrenal, adenoma parotídeo y carcinoma neuroendocrino.
2.3.- Seguimiento
La evaluación inicial tras el diagnóstico será: un examen dermatológico y biopsia de lesiones cutáneas sospechosas, TC tórax para visualizar quistes pulmonares (basal) y RM o TAC o ecografía abdominal, cada año.
3.- Lleiomiomatosis Múltiple y Cáncer Renal Hereditarios
Es una genodermatosis, con muy baja incidencia, <500 familias en todo el mundo, por lo que se considera una enfermedad rara. Su herencia es autosómica dominante, se debe a mutaciones en el gen de la fumarato hidratasa, FH, localizado en el cromosoma 1q42.3-43. La enzima fumarato hidratasa participa en el ciclo de Krebs.
3.2.- Características clínicas
Se caracteriza por la aparición de leiomiomas cutáneos múltiples (a partir del músculo erector del pelo), leiomiomas uterinos en las mujeres, quistes renales y cáncer renal.
3.2.1.- Leiomiomas cutáneos
Aparece en un 85% de los pacientes. Son tumores benignos originados en el músculo piloerector del folículo piloso y se manifiestan con pápulas y nódulos de superficie lisa, brillante de color marrón o rojizo. El tamaño es de entre 0,2 y 2,5 cm y el número es variables, desde 1 a >100. Las lesiones pueden aumentar con el tiempo.
Suelen debutar a los 25 años, si bien pueden aparecer desde la adolescencia hasta la 4ª década de la vida. Generalmente se localizan en tronco y extremidades. Presentan una distribución agrupada, diseminada o segmentaria. Las lesiones de mayor tamaño pueden ser dolorosas a la presión, al frío o al calor. La transformación maligna es rara.
En ocasiones requieren tratamiento mediante escisión quirúrgica, láser o crioablación.
3.2.2.- Leiomiomas uterinos
Son lesiones benignas, presentes en un 25-77% de mujeres durante la edad fértil. Debutan con dolor abdominal, sangrado menstrual intenso, infertilidad o como hallazgo casual en piezas de histerectomía.
La edad de aparición media es de 30 años, más precoz que en la población general (40-45 años). Suelen ser tumores múltiples y de gran tamaño (10 cm). El 91-98% de las mujeres con leiomiomas cutáneos desarrollan leiomiomas uterinos.
Se han descrito casos aislados de transformación maligna a leiomiosarcomas. Revisiones recientes confirman que son leiomiomas atípicos, pero no leiomiosarcomas.
Los leiomiomas uterinos pueden ser tratados quirúrgicamente mediante miomectomía o histerectomía en mujeres de riesgo y sin deseos reproductivos o mediante tratamiento médico con agonistas LHRH o tratamientos anti·hormonales.
3.2.3.- Tumores renales
Presentes hasta en un 20% de las familias diagnosticadas de leiomiomatosis hereditaria, a una edad media de 44 años, pero puede presentarse a edades muy jóvenes, con 20-30 años. Se consideraba que eran exclusivamente de subtipo carcinoma papilar tipo 2, pero también pueden ser de subtipo tubulocístico, o incluso de subtipo células claras. Generalmente son unilaterales, solitarios y metastáticos al diagnóstico. Es la principal causa de mortalidad. Por ello, se recomienda el tratamiento quirúrgico mediante nefrectomía total, incluso en estadios precoces.
3.3.- Manejo de portadores
Se recomienda realizar una revisión dermatológica y ginecológica con ecografía transvaginal, cada 12 meses. Para el cribado de los tumores renales se indica realizar una TC o RM abdominal cada 12 meses.
4.- Carcinoma renal papilar hereditario
La incidencia es desconocida y hay pocas familias descritas.
Presenta una herencia autosómica dominante, con una gran penetrancia, aunque incompleta, con una probabilidad de desarrollar carcinoma de células renales hasta del 90% a los 80 años.
4.1.- Genética
El carcinoma papilar del riñón se asocia a mutaciones en línea germinal en el oncogen MET, localizado en el cromosoma 7q31.
4.2.- Rasgos Clínicos
Es más frecuente en varones (x2.4). La edad media del diagnóstico son los 45 años. Se caracteriza por la aparición de múltiples tumores renales bilaterales papilares de tipo 1. Suelen diagnosticarse como un incidentaloma al realizar una TAC o debutar con hematúria, dolor abdominal y masa renal. Presentan una lenta evolución, con un potencial metastásico menor que los papilares tipo 2 asociados a Leiomiomatosis Múltiple y Cáncer Renal Hereditario.
4.3.- Manejo
Se recomienda realizar una prueba de imagen abdominal (TC o RM) cada 12 meses.
5.- Complejo Esclerosis Tuberosa (CET)
El CET es un trastorno neuro·cutáneo con afectación sistémica: riñones, pulmón, ojos, corazón e hígado; en el cual aparecen una gran variedad de tumores benignos, susceptibles de malignizar. Su incidencia es de 1 de cada 6000 recién nacidos vivos y 1 de cada 8000 adultos.
5.1.- Genética
Se asocia a mutaciones en el gen TSC1 y TSC2, localizados en el cromosoma 9p34 y 16p13.3, respectivamente. Presenta una herencia autosómica dominante con un fenotipo muy variado.
El gen TSC1 codifica la proteína hamartina y el gen TSC2 la proteína tuberina. El complejo tuberina·hamartina está implicado en el crecimiento celular a través de la síntesis proteica.
5.2.- Clínica
El diagnóstico clínico clásicamente se había realizado por la tríada de Vogt, presente en menos de 1/3 de los pacientes: epilepsia, retraso mental y adenomas sebáceos.
5.2.1.- Manifestaciones cutáneas
Las lesiones cutáneas más características son las lesiones “en hoja de fresno” que suelen aparecer en el tronco, glúteos y escápula (90-95%) y los angiofibromas bilaterales en la región malar (80%). También pueden aparecer placas fibrosa marrones, placas de Shagreen, fibromas ungueales, fibromas gingivales y alteraciones de la dentición.
Las lesiones cutáneas aumentan en tamaño y número hasta la pubertad.
Para prevenir la aparición de lesiones cutáneas se recomienda fotoprotección solar. El tratamiento quirúrgico de las lesiones cutáneas se indica por razones estéticas, por la aparición de síntomas o un rápido crecimiento de las lesiones.
5.2.2.- Manifestaciones neurológicas
Un 90% de los niños diagnosticados de CET presentan hamartomas glio·neuronales y/o nódulos subependimarios que provocan crisis epileptiformes refractarias a tratamiento (63%), déficit cognitivo y trastornos conductuales del espectro autismo. También pueden aparecer lesiones desmielinizantes y astrocitomas subependimarios de células gigantes, neoplasia benigna de bajo potencial de malignización.
El tratamiento quirúrgico de los tumores cerebrales se indicará cuando las lesiones provoquen sintomatología. Aquellos tumores considerados irresecables o inoperables por comorbilidad pueden beneficiarse de tratamiento con everolimus.
5.2.3.- Manifestaciones cardio·vasculares
Un 58% de las CET presentan rabdomiomas cardíacos, lesiones tumorales benignas sin capacidad de malignización, que suelen degenerar espontáneamente. Pero con capacidad de morbi·mortalidad derivada de arritmias e infarto agudo de miocardio.
5.2.4.- Manifestaciones renales
El 80% de los pacientes presentarán lesiones renales: angiomiolipomas (49%), quistes renales (26%), carcinoma de células renales (2,3%), oncocitomas, enfermedad intersticial renal o glomerulonefritis focal y segmentaria.
Los angiomiolipomas son neoplasias con riesgo de malignización. El tratamiento quirúrgico se indicará principalmente cuando su tamaño supere los 4 cm, provoquen síntomas o ante la sospecha de malignización.
El carcinoma de células renales presenta un debut precoz, pudiendo aparecer incluso en la infancia. Se caracteriza por ser multifocal y bilateral. Por ello, el tratamiento quirúrgico será conservador.
5.2.5.- Manifestaciones oftalmológicas
Las manifestaciones oftalmológicas pueden ser: hamartomas retinianos (44%), parches retinianos acrómicos, angiofibromas del párpado (39%), colobomas, despigmentación del iris, estrabismo paralítico y alteraciones de la refractariedad.
5.3.- Manejo clínico del CET
Se recomienda una inspección oral al diagnóstico, exploración dermatológica, neurológica y oftalmológica anual. Así como la monitorización de la función renal mediante el aclaramiento de creatinina, una TC torácica cada 12 meses y una RM abdominal cada 1-3 años.
Tabla 1: Síndromes de cáncer hereditario más frecuentes asociados a carcinoma renal

Tabla 2: Seguimiento en los pacientes diagnosticados de VHL


