


Sarcomas óseos
Índice de navegación
- Sarcomas óseos
- Centros de referencia en sarcomas (CSUR)
- Tipos de sarcomas óseos
- Frecuencia de los sarcomas óseos
- Proceso diagnóstico ante un posible sarcoma óseo
- Cirugía y quimioterapia en el tratamiento de sarcomas del hueso
- Diagnóstico, pronóstico y tratamiento de los Osteosarcomas
- Diagnóstico, pronóstico y tratamiento del sarcoma de Ewing esquelético
- Otros tumores oseos
- Todas las páginas
AUTORA:
Dra. Gloria Marquina
Introducción
Los sarcomas son un conjunto de cánceres raros mesenquimales (en torno 1.5-2% de todos los cánceres) que se diferencian hacia tejido muscular, tejido óseo, tejido nervioso, tejido cartilaginoso, tejido tendinoso, tejido de vasos sanguíneos, tejido adiposo…
Pueden diagnosticarse a cualquier edad y en cualquier localización, siendo la más frecuente en las extremidades, seguida de tronco, retroperitoneo, cabeza y cuello, visceral…
Existen más de 70 subtipos de sarcomas y se dividen en tres grandes entidades: sarcomas de partes blandas, sarcomas óseos y tumores del estroma gastrointestinal (GIST). La última actualización de la Organización Mundial de la Salud (OMS, WHO por sus siglas en inglés) de los sarcomas óseos se ha publicado en 2020.
Es fundamental destacar la existencia de centros de referencia para pacientes con sarcomas en España (CSUR) desde el año 2017.
En esta información nos centraremos en los sarcomas óseos.
Centros de referencia en sarcomas (CSUR)
La creación en España de centros de referencia en sarcomas ha supuesto un avance para los pacientes con sarcomas. Actualmente contamos con siete centros de referencia en sarcomas en España.
La razón de ser de los CSUR de sarcomas en España es centralizar la atención de los pacientes con sarcomas para brindar al paciente un abordaje multidisciplinar diagnóstico-terapéutico y, por ende, impactar positivamente en la supervivencia de los pacientes con sarcomas. En España, la derivación de pacientes a centros de referencia no es obligatoria tal y como ocurre en otros países como Reino Unido o Francia, pero es un gran avance para los pacientes con sarcoma que nos sitúa cerca de la situación de otros países de nuestro entorno.
Puede encontrar el listado de CSUR en el siguiente enlace web en la página 12: https://www.sanidad.gob.es/profesionales/CentrosDeReferencia/docs/ListaCSUR.pdf
Tipos de sarcomas óseos
La clasificación de los tumores malignos primarios del hueso contempla 8 grandes grupos: osteosarcoma, condrosarcoma, sarcoma de Ewing, angiosarcoma, fibrosarcoma, cordoma, adamantimoma y otros sarcomas. Se excluyen los tumores derivados de las células de la sangre (linfomas, mieloma) así como las metástasis de tumores de otro origen.
En este capítulo vamos a tratar principalmente los dos tipos de sarcomas óseos más predominantes: el osteosarcoma convencional y el sarcoma de Ewing.
Frecuencia de los sarcomas óseos
Los sarcomas óseos son tumores muy infrecuentes y afectan sobre todo a niños y a adolescentes. La incidencia se estima en 1 paciente nuevo por año por cada 100000 habitantes. A pesar de su rareza, constituyen la quinta causa de cáncer en adolescentes y jóvenes entre 15 a 19 años. En estos pacientes, algo más de la mitad de los sarcomas óseos son osteosarcomas, una tercera parte son sarcomas de Ewing y menos del 10% son condrosarcomas. El sarcoma de Ewing es muy poco frecuente en adultos. Sin embargo, el osteosarcoma tiene una distribución por edades de tipo bimodal, esto es, con dos momentos de mayor frecuencia: uno entre los 13 a 16 años y el otro por encima de los 65 años.
Proceso diagnóstico ante un posible sarcoma óseo
Los síntomas más frecuentes de los sarcomas óseos son dolor y/o aparición de una tumoración en una localización ósea. Habitualmente los síntomas son de meses o, incluso, años de instauración. Es frecuente que exista un antecedente de traumatismo o sobrecarga ósteo-muscular, aunque no como causa, sino como desencadenante de los síntomas.
La primera técnica diagnóstica que se debe realizar es una radiografía simple de hueso. Esta técnica es fácilmente accesible y puede dar mucha información para el diagnóstico diferencial de la lesión ósea.
Tras la radiografía, se debe completar el estudio mediante una tomografía axial computerizada (TAC) y/o Resonancia Magnética Nuclear (RMN).

Figura 1. Radiología simple de rodilla anteroposterior: osteosarcoma en cóndilo femoral medial izquierdo.
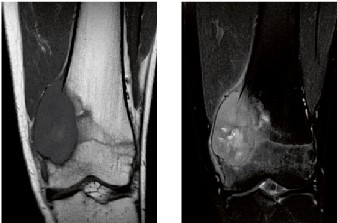
Figura 2. Resonancia Magnética Nuclear: cortes coronales rodilla izquierda con imágenes potenciadas en T1 (izquierda) e imagen potenciada en T2 con saturación grasa (izquierda). Osteosarcoma en cóndilo femoral medial izquierdo.
Para el diagnóstico definitivo del sarcoma óseo, se requiere de la obtención de una biopsia de tejido tumoral. La planificación de la biopsia es esencial para un buen resultado terapéutico. Para este fin pueden usarse técnicas radiológicas, habitualmente la TAC, para guiar la trayectoria de dicha aguja. La biopsia se debe plantear en comité multidisciplinar de sarcomas y consensuando el trayecto con el equipo quirúrgico que va a tratar al paciente. Se recomienda que sea realizada en centros de referencia en sarcomas o centros que cuenten con equipos multidisciplinares de sarcomas.
Finalmente, se debe realizar un estudio de extensión para descartar metástasis. Para ello se utiliza TAC de tórax-abdomen – para descartar metástasis pulmonares-, gammagrafía ósea – para descartar afectación de otros huesos del esqueleto o PET-TC de cuerpo entero.
Generalidades de la cirugía y la quimioterapia en el tratamiento de los sarcomas del hueso
El tratamiento de los sarcomas óseos es multidisciplinar, lo que quiere decir que requiere la intervención de varios especialistas. La cirugía habitualmente constituye un componente esencial del dicho tratamiento. El cirujano debe estar involucrado en el diseño de la mejor estrategia de toma de biopsia, como hemos visto previamente.
El procedimiento quirúrgico específico depende de la localización y de la extensión del tumor primario.

Figura 3. Radiografía simple: Cirugía conservadora de extremidad: megaprótesis en fémur izquierdo (cirugía de osteosarcoma).
La cirugía conservadora de miembro persigue la exéresis completa del tumor, con márgenes de seguridad, así como la sustitución del defecto estructural mediante la implantación de prótesis o injertos (ver un ejemplo en la Figura 3). Cuando el tumor es muy voluminoso, algunos estudios han mostrado que la administración de quimioterapia pre-operatoria puede aumentar la tasa de cirugías no mutilantes. En cualquier caso, la cirugía conservadora se hará siempre que se pueda garantizar la exéresis completa, y que su realización no reste probabilidades de supervivencia. Aunque la amputación de extremidad no es algo deseable, con los avances de hoy día en prótesis ortopédicas muy funcionales los pacientes amputados pueden lograr una calidad de vida muy funcional y plenamente integrada.
En algunos casos de recaída local o de metástasis también puede plantearse la cirugía de rescate en comité multidisciplinar, individualizando caso por caso.
Las consideraciones sobre los efectos secundarios de la quimioterapia se recogen en otra parte de las guías de la SEOM para pacientes. No obstante, es importante conocer que la mayor parte de los esquemas quimioterapéuticos utilizados en el tratamiento los sarcomas óseos va a requerir antieméticos (fármacos para reducir el riesgo de náuseas y vómitos) y factores estimuladores de las células precursoras de los glóbulos blancos. Hay varios fármacos potencialmente útiles para este fin, agrupados bajo el acrónimo inglés G-CSF, que quiere decir factor que estimula las colonias de granulocitos (un tipo de glóbulo blanco). Ocasionalmente serán necesarias transfusiones de derivados de la sangre (glóbulos rojos, plaquetas). En pacientes con anemia inducida por quimioterapia debe estudiarse la necesidad de recibir tratamiento con eritropoyetina.
Hay que abordar una posible infertilidad futura antes de iniciar la quimioterapia. Remitimos a la guía de la SEOM que enfoca el manejo de esta toxicidad.
Tras el fin de todos los tratamientos, el paciente debe ser evaluado periódicamente por el equipo multidisciplinar, no sólo para descartar la recidiva del sarcoma óseo, sino para evaluar posible toxicidad a largo plazo de los tratamientos utilizados.
Aspectos específicos del diagnóstico, pronóstico y tratamiento de los Osteosarcomas
Las causas exactas del osteosarcoma no son conocidas. El desarrollo de un osteosarcoma puede estar en relación con el crecimiento del hueso rápido, por ejemplo, durante la pubertad. Esto podría explicar la aparición, sobre todo, en adolescentes.
La presencia de enfermedad de Paget o el haber recibido radioterapia sobre el hueso en el pasado puede implicar un mayor riesgo de padecer un osteosarcoma en el futuro.
Muy raramente el osteosarcoma puede ocurrir como consecuencia de una alteración genética, como en el caso del Síndrome de Li-Fraumeni.
Es esencial que el diagnóstico y el tratamiento se realicen en centros en los que exista experiencia en el abordaje multidisciplinar de esta enfermedad.
Aspectos específicos del diagnóstico
A la hora de realizar el diagnóstico de un osteosarcoma, hay que determinar:
- Estadio: si es localizado o metastásico.
- Grado: bajo, moderado o alto, en función de la apariencia de las células si son muy parecidas al hueso normal (bajo grado) o muy alteradas respecto al hueso normal (alto grado). La mayoría de los osteosarcomas son de alto grado.
Aspectos específicos del tratamiento
Enfermedad localizada
El tratamiento del osteosarcoma localizado se basa en cirugía y quimioterapia.
El esquema de tratamiento de quimioterapia que se utiliza en osteosarcoma es:
- < 30 años: MAP – Metotrexate a altas dosis, Adriamicina, Cisplatino.
- >30 años: Adriamicina y Cisplatino.
Se utiliza la quimioterapia pre-operatoria antes de la cirugía (neoadyuvante), cirugía y quimioterapia postoperatoria (adyuvante). La adición de otros fármacos como Ifosfamida no ha mejorado los resultados en supervivencia.
La presencia de necrosis en más del 90% del tumor en el análisis de la pieza resecada en la cirugía demuestra una adecuada quimiosensibilidad y se asocia a mejor pronóstico del osteosarcoma.
Como dato importante, metotrexate a dosis altas es un tratamiento que requiere hospitalización puesto que se administran sueros y bicarbonato intravenosos antes y después de la infusión del fármaco de cara a alcalinizar la orina para que no cristalice el metotrexate en el interior del riñón. Además, hay que medir los niveles de metotrexate en sangre hasta estar seguros de que bajan adecuadamente por debajo del umbral de toxicidad. A las 24 horas de la infusión de metotrexate a altas dosis, hay que administrar un antídoto denominado ácido folínico (Leucovorin), que sirve para disminuir la probabilidad de que ocurra una toxicidad grave.
En pacientes menores de 30 años, la adición de un agente inmunomodulador en el tratamiento adyuvante, la mifamurtida, es capaz de incrementar la supervivencia de pacientes con osteosarcoma de alto grado localizado.
Enfermedad metastásica
Se debe determinar si la enfermedad metastásica es potencialmente resecable. Si es así, el tratamiento de la enfermedad metastásica seguirá el mismo esquema de la enfermedad localizada.
Cuando sólo haya metástasis pulmonares, la cirugía de las mismas se puede considerar potencialmente curativa, ya que puede lograr largos periodos de remisión de la enfermedad y en casos seleccionados, incluso la curación.
En caso de que la enfermedad metastásica no sea potencialmente resecable, se administrará tratamiento con quimioterapia y se valorará en comité multidisciplinar el realizar cirugía del tumor primario.
Enfermedad recurrente
Es fundamental valorar en comité multidisciplinar si la recaída es resecable quirúrgicamente. No está bien determinado el papel de la quimioterapia post-cirugía en estos casos.
En caso de que no sea resecable, el paciente recibirá tratamiento con quimioterapia. Se recomienda ofrecer al paciente la participación en ensayos clínicos.
Aspectos específicos del diagnóstico, pronóstico y tratamiento del sarcoma de Ewing esquelético
El sarcoma de Ewing es el representante más destacado de una familia de tumores denominada, también, de Ewing. Los otros tumores de este grupo son el tumor neuroectodérmico periférico primitivo (PNET), el sarcoma de Ewing extra-esquelético, el neuroblastoma del adulto, el tumor maligno de célula pequeña de la región tóraco-pulmonar (tumor de Askin), el tumor paravertebral de célula pequeña y el sarcoma de Ewing atípico.
A continuación, nos centraremos en el sarcoma de Ewing óseo.
No se tiene conocimiento sobre las causas exactas del desarrollo del sarcoma de Ewing óseo. Los tumores de la familia del sarcoma de Ewing se caracterizan por tener anomalías cromosómicas que implican a los cromosomas 11 y 22. Como consecuencia de estas anomalías cromosómicas se genera una proteína anormal. En la actualidad se está investigando el significado biológico y el valor pronóstico de estas proteínas.
Aspectos específicos del diagnóstico
El sarcoma de Ewing óseo tiene predilección por los huesos largos de las extremidades o por los de la pelvis.
La mayoría de las metástasis ocurren en el pulmón o en los huesos. Dentro de los huesos, el sarcoma de Ewing tiene especial predilección por los de la columna.
El sarcoma de Ewing puede dar lugar a metástasis en médula ósea por lo que el estudio de extensión se debe completar con la realización de una biopsia/aspirado de médula ósea aunque hay datos cada vez más consistentes en que la realización de una PET-TAC como estudio de extensión puede ser más sensible que la biopsia de médula ósea para descartar metástasis en médula ósea.
Las técnicas moleculares para detectar la proteína de fusión EWS/FLI resultante de la fusión de los cromosomas 11 y 22, son imprescindibles para el diagnóstico de sarcoma de Ewing.
Aspectos específicos del pronóstico
Se deben tener en cuenta:
- Extensión de la enfermedad: ausencia o presencia de metástasis.
- La afectación metastásica conlleva un peor pronóstico. Dentro de este grupo, los pacientes que sólo tienen metástasis pulmonares evolucionan mejor que los que tienen metástasis óseas o en ambos órganos.
- La localización y el tamaño tumoral:
- La afectación de huesos del esqueleto axial (columna, pelvis, cráneo) tiene peor pronóstico que la afectación de huesos de las extremidades.
- Los pacientes con tumores primarios pequeños (con un volumen estimado de menos de 100 centímetros cúbicos) sobreviven más que los pacientes con tumores de mayor volumen. Así mismo, los parámetros relacionados con el tamaño tumoral (anemia, fiebre, elevación de la LDH) se relacionan también con peor pronóstico.
- Respuesta a tratamiento:
- Si se detecta un porcentaje elevado de tumor viable en el tumor resecado en la cirugía tras administración de quimioterapia, el pronóstico es peor que si no se detectan células tumorales o si sólo queda una enfermedad residual mínima.
- Edad: este factor es controvertido. En niños hay datos que apoyan que el pronóstico es mejor a menor edad. Sin embargo, otros autores interpretan estos datos como la consecuencia de que en los adultos sea más frecuente encontrar tumores más grandes que en los niños, así como que la tolerancia a la quimioterapia puede ser peor.
Aspectos específicos del tratamiento
Tratamiento de la enfermedad localizada
El tratamiento del Sarcoma de Ewing localizado consiste en quimioterapia (neoadyuvante), antes de la realización del tratamiento loco-regional con cirugía y/o radioterapia y quimioterapia posterior al tratamiento loco-regional (adyuvante).
Los esquemas de quimioterapia que se utilizan habitualmente son:
- VIDE: Vincristina, Ifosfamida, Doxorrubicina, Etoposido.
- VDC/IE: Vincristina, Doxorrubicina, Ciclofosfamida, alternando con Ifosfamida y Etoposido. En pacientes jóvenes con enfermedad localizada se ha podido demostrar mayor beneficio si la quimioterapia se administra cada dos semanas en lugar de cada tres.
El tratamiento con quimioterapia a altas dosis seguido de trasplante de células progenitoras hematopoyéticas (células de la sangre) es controvertido en la actualidad en Sarcoma de Ewing.
Tratamiento locorregional: cirugia y/o radioterapia
El control local de la enfermedad se puede conseguir mediante cirugía, radioterapia o ambos. Cada uno tiene sus ventajas y sus inconvenientes. Lo recomendable es el tratamiento dentro de un equipo multidisciplinar con experiencia en esta enfermedad.
El sarcoma de Ewing es un tumor radiosensible. La radioterapia puede constituir una elección óptima en pacientes en los que el tratamiento quirúrgico sea mutilante o genere secuelas de pérdida de función.
En aquellos casos en los que la cirugía no consiga una resección completa de la enfermedad, con márgenes no afectos (esto es, no infiltrados por el tumor), puede contemplarse la indicación de radioterapia postquirúrgica. En centros especializados se puede contemplar la opción de la radioterapia intraoperatoria, en el propio acto quirúrgico, tras la extirpación de tumores grandes o con dificultad de exéresis completa.
Tratamiento de la enfermedad metastásica al diagnóstico
Se utilizan los mismos esquemas de tratamiento de quimioterapia de la enfermedad localizada y se intenta control local de la enfermedad y/o de la enfermedad metastásica. Algunos pacientes con enfermedad metastásica en el momento del diagnóstico pueden curarse. Se debe individualizar el tratamiento en estos pacientes en comité multidisciplinar.
Tratamiento de la enfermedad recurrente
La mayoría de las recaídas ocurren durante los primeros 5 años, aunque pueden detectarse a largo plazo, por lo que el seguimiento de estos pacientes debe ser prolongado.
El tratamiento habitual es quimioterapia. Se valorará en comité multidisciplinar la posibilidad de realizar tratamiento quirúrgico o radioterápico de la enfermedad recurrente.
Se recomienda ofrecer al paciente la participación en ensayos clínicos.
Complicaciones a largo plazo de los tratamientos
No hay muchos datos acerca de los efectos a largo plazo de los diversos tratamientos contra el sarcoma de Ewing. Están descritas las fracturas patológicas sobre el hueso tratado. Estas complicaciones cada vez son menores debido al avance en los últimos años de las técnicas de radioterapia (complicaciones de la herida quirúrgica, fibrosis pulmonar, neuropatía, asimetría de la longitud de las extremidades, necrosis de la cabeza del fémur). La quimioterapia también puede dejar secuelas en algunos casos, como pueden ser insuficiencia renal, toxicidad intestinal, neuropatía, disfunción cardiaca.
Se recomienda la lectura de las páginas de la SEOM destinadas a los efectos secundarios de los fármacos antineoplásicos.
Otros tumores óseos
Principales características de otros sarcomas óseos relativamente frecuentes:
- Condrosarcoma: Es un tumor más frecuente (0.2 casos por 100000 habitantes). En pacientes adultos tiene poca tendencia a producir metástasis a distancia. En la mayor parte de los casos el pronóstico es bastante favorable, y el tratamiento se basa en la cirugía. Cabe destacar que en la última actualización de la WHO de 2020, en función de dónde se localice el tumor de estirpe cordal se puede denomidar tumor cartilaginoso atípico (si está localizado en extremidades) o condrosarcoma ( si está localizado en tronco).
- Cordoma: Tumor maligno que afecta principalmente a la columna vertebral, especialmente a nivel sacro y cervical. Es un tumor de lento crecimiento, pero con gran tendencia a la recidiva local y con capacidad metastatizante tras múltiples recaídas. El tratamiento se basa en la cirugía inicial, pero en muchos casos es difícil lograr resecciones completas. Es una cirugía compleja y muy especializada que debe ser realizada en centros de referencia. La radioterapia puede ser también un tratamiento eficaz, en especial la radioterapia con protones (actualmente está en marcha un ensayo clínico “SACRO” de protonterapia vs cirugía en centro experto reclutando pacientes en Europa).
- Tumor de células gigantes del hueso: Suele afectar a huesos largos en pacientes de mediana edad. Tiene gran tendencia a recidivar e incluso producir metástasis en el pulmón. En el 8% de los casos esta descrito que puede malignizar hacia sarcomas más agresivos. Además de la cirugía, el tratamiento con Denosumab (anticuerpo monoclonal dirigido contra un receptor que expresa el tumor - RANKL) ha demostrado eficacia en la enfermedad metastásica y en neoadyuvancia, para ayudar a realizar una cirugía conservadora en el paciente.

